Противовирусные агенты. I. Синтез 1- [-(3, 5-диметилфенокси) алкил]-производных урацила
![Противовирусные агенты. I. Синтез 1- [-(3, 5-диметилфенокси) алкил]-производных урацила](/file/picture/142148890/protivovirusnye-agenty-i-sintez-13-5-dimetilfenoksi-alkilproizvodnyh.png "Противовирусные агенты. I. Синтез 1- [-(3, 5-диметилфенокси) алкил]-производных урацила")
Автор: Бабков Д.А., Парамонова М.П., Озеров А.А., Новиков М.С.
Журнал: Волгоградский научно-медицинский журнал @bulletin-volgmed
Рубрика: Фармакология токсикология
Статья в выпуске: 2 (30), 2011 года.
Бесплатный доступ
Конденсацией эквимолярных количеств 2,4-бис(триметилсилилокси)пиримидина и 1-бром-ω-(3,5-диметилфенокси)алканов был осуществлен синтез 1-[ω-(3,5-диметилфенокси)алкил]-производных урацила, выход которых составил 68-80 %. Изучены физико-химические и спектральные свойства синтезированных соединений. Данные соединения представляют интерес в качестве потенциальных анти-ВИЧ-1 агентов.
Синтез, урацил, 4-бис(триметилсилилокси)пиримидины, потенциальные противовирусные агенты
Короткий адрес: https://sciup.org/142148890
IDR: 142148890 | УДК: 615.3:547.854.4
Antiviral agents. I. Synthesis of 1- [-(3, 5-dimethylphenoxy) alkyl]-derivatives of uracil
Condensation of equimolar quantities of 2,4-bis(trimethylsilyloxy)pyrimidine and 1-bromo-ω-(3,5-dimethylphenoxy)alkanes led to 1-[
Текст научной статьи Противовирусные агенты. I. Синтез 1- [-(3, 5-диметилфенокси) алкил]-производных урацила
С момента открытия вируса иммунодефицита человека (ВИЧ) в качестве этиологического агента синдрома приобретенного иммунодефицита (СПИД) до настоящего времени ВИЧ-инфекция остается одной из наиболее серьезных клинических проблем. В комплексной терапии ВИЧ-инфекции применяются три группы препаратов: нуклеозидные (НИОТ) и ненуклеозидные (ННИОТ) ингибиторы обратной транскриптазы и ингибиторы протеазы [3].
Ингибиторы ОТ ВИЧ позволяют подавить репликацию вируса на ранних стадиях жизненного цикла. Среди последних именно ненуклеозидные ингибиторы являются наиболее перспективными [6]. В отличие от НИОТ, они не встраиваются в метаболические пути клетки, обладают высокой специфичностью действия за счет аффинитета к аллостерическому центру фер- мента, характеризуются значительно меньшей токсичностью. Однако эффективность используемых в настоящее время ННИОТ (невирапин, делавердин, эфа-виренз) серьезно ограничена неудовлетворительными фармакокинетическими профилями препаратов, а главное – быстрой селекцией резистентных к ним мутантных штаммов ВИЧ [5]. Поэтому поиск новых ингибиторов репликации ВИЧ, потентных как в отношении дикого штамма, так и клинически важных изолятов вируса, является чрезвычайно актуальной задачей.
Среди соединений, являющихся ациклическими аналогами пиримидиновых нуклеозидов, описаны многие, обладающие выраженной биологической активностью, в том числе противовирусной и цитостатической. Ранее сообщалось о некоторых соединениях этого ряда, продемонстрировавших значи- тельную анти-ВИЧ активность in vitro [1]. Было также показано, что наличие участка с высокой электронной плотностью в конце ациклической цепи является необходимым условием для появления вирус-инги-биторных свойств в соединениях этого типа.
ЦЕЛЬ РАБОТЫ
Разработать методы синтеза, исследовать физико-химические и спектральные свойства ациклических аналогов пиримидиновых нуклеозидов, содержащих в положении N1 пиримидинового цикла алкильный заместитель с терминальным 3,5-диме-тилфенокси-фрагментом.
МЕТОДИКА ИССЛЕДОВАНИЯ
Спектры ЯМР 1Н регистрировались на спектрометре «BrukerAvance 400» (400 МГц) в ДМСО-D6, внутренний стандарт – тетраметилсилан. Тонкослойная хроматография выполнена на пластинах «Сорбфил» (Россия), элюент – этилацетат, проявление в парах йода. Температуры плавления измерены в стеклянных капиллярах на приборе «Mel-Temp 3.0» (Laboratory Devices Inc., США).
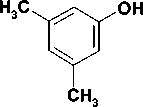
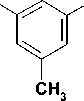
1-бром-3-(3,5-диметилфенокси)пропан (1) . В круглодонную колбу на 0,5 л помещают 15 г (0,1228 моль) 3,5-диметилфенола, 25,5 г (0,1842 моль) прокаленного карбоната калия и 100 мл метилэтилкето-на, добавляют 51,4 мл (0,4912 моль) 1,3-дибромпропа-на, осторожно кипятят с обратным холодильником в течение 20 часов. Осадок отфильтровывают, промывают на фильтре этилацетатом (3 х 10 мл), фильтрат упаривают при пониженном давлении, остаток перегоняют в вакууме, собирая фракцию, кипящую при 141—144 оС (5 мм рт. ст.). Получают 18,51 г (выход 62 %) бромида 1 в виде бледно-желтой вязкой жидкости.
Соединения 2-4 были получены аналогично.
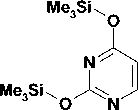
1-[3-(3,5-диметилфенокси)пропил]урацил (5). К 2,4-бис(триметилсилилокси)пиримидину, полученному кипячением 1,5 г (13,4 ммоль) урацила в 50 мл ГМДС в присутствии каталитического количества хлорида аммония, добавляют 3,26 г (13,4 ммоль) 1 и нагревают при 180—190 оС в течение 30 мин с защитой воздуха от влаги. Образовавшуюся вязкую прозрачную светло-коричневую массу оставляют на ночь при комнатной тем- пературе. Растворяют в 25 мл этилацетата и 10 мл изопропанола. Выделившийся осадок отфильтровывают, промывают на фильтре этилацетатом (2 х 10 мл), сушат на воздухе при комнатной температуре и дважды кристаллизуют из смеси 30 мл изопропанола и 10 мл ДМФА. Получают 2,5 г соединения 8 (выход 68 %) в виде белого кристаллического вещества. 1H ЯМР-спектр (ДМСО-D6), δ, м.д., J (Гц): 1,95 т (2H, J = 6,3, CH2); 2,15 с (6H, CH3); 3,77 т (2H, J = 6,0, NCH2); 3,87 т (2H, J = 5,7, OCH2); 5,48 д (1H, J = 7,5, H-5); 6,44 с (2H, H-2’, H-6’); 6,50 с (1H, H-4’); 7,55 д (1H, J = 8,1, H-6); 11,20 с (1H, NH).
Соединения 6-8 были получены аналогично.
1-[4-(3,5-диметилфенокси)бутил]урацил (6) .
1H ЯМР-спектр (ДМСО-D6), δ , м.д., J (Гц): 1,63 с (4H, CH2, CH2); 2,15 с (6H, CH3); 3,66 т (2H, J = 6,2, NCH2); 3,86 т (2H, J = 6,2, OCH2); 5,50 дд (1H, J = 7,8 и 2,1, H-5); 6,46 с (2H, H-2’, H-6’); 6,49 с (1H, H-4’); 7,61 д (1H, J = 7,5, H-6); 11,20 с (1H, NH).
1-[5-(3,5-диметилфенокси)пентил]урацил (7) .
1H ЯМР-спектр (ДМСО-D6), δ , м.д., J (Гц): 1,37 к (2H, J = 6,8, CH2); 1,62 к (2H, J = 7,2, CH2); 1,67 к (2H, J = 7,2, CH2); 2,21 с (6H, CH3); 3,66 т (2H, J = 7,1, NCH2); 3,88 т (2H, J = 6,4, OCH2); 5,55 дд (1H, J = 7,9 и 2,2, H-5); 6,51 с (2H, H-2’, H-6’); 6,53 с (1H, H-4’); 7,64 д (1H, J = 7,8, H-6); 11,25 с (1H, NH).
1-[6-(3,5-диметилфенокси)гексил]урацил (8) .
1H ЯМР-спектр (ДМСО-D6), δ , м.д., J (Гц): 1,23 м (2H, J = 6,6, CH2); 1,36 м (2H, J = 6,9, CH2); 1,53 м (2H, J = 7,3, CH2); 1,62 м (2H, J = 5,4, CH2); 2,15 с (6H, CH3); 3,59 т (2H, J = 7,4, NCH2); 3,83 т (2H, J = 6,5, OCH2); 5,47 дд (1H, J = 8,0 и 1,8, H-5); 6,46 с (2H, H-2’, H-6’); 6,48 с (1H, H-4’); 7,59 д (1H, J = 8,1, H-6); 11,17 с (1H, NH).
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
И ИХ ОБСУЖДЕНИЕ
Целевые соединения были синтезированы в несколько этапов. Бромиды 1-4 были получены по реакции Вильямсона (рис.) нагреванием 3,5-диметилфенола с 4-кратным мольным избытком терминальных дибромалканов и 1,5-кратным избытком безводного карбоната калия в среде метилэтилкетона в соответствии с известными методами [4]. При этом выход бромидов 1-4 составил 62—74%.
Br(CH 2 ) n Br, K 2 CO 3
CH3COC2H5
180-190 oC
H 3 C
1-4
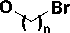
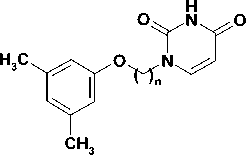
5-8
Рис. Схема синтеза 1-[ ω -(3,5-диметилфенокси)]алкильных производных урацила
Затем 2,4-бис(триметилсилилокси)пиримидин, полученный нагреванием урацила с избытком гексаметилдисилазана в присутствии хлорида аммония, конденсировали с полученными бромидами 1-4 по методу Гилберта-Джонсона, что позволило с высокой селективностью вводить заместитель в положение N1 [2]. Выход целевых соединений 5-8 составил 68—80 %.
Чистота полученных соединений ( 5-8 ) определялась методом тонкослойной хроматографии, строение подтверждено данными 1Н ЯМР-спектроскопии, их физико-химические свойства представлены в табл.
Свойства синтезированных соединений
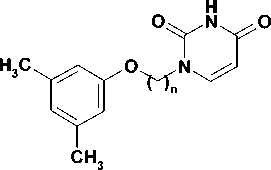
|
Соединение |
n |
R f * |
Т пл , °С |
Выход, % |
|
5 |
2 |
0,49 |
89,5 — 91 |
71 |
|
6 |
3 |
0,43 |
152 — 153 |
68 |
|
7 |
4 |
0,58 |
96 — 98 |
80 |
|
8 |
5 |
0,54 |
130 — 131,5 |
76 |
* Элюент — этилацетат.
ЗАКЛЮЧЕНИЕ
Таким образом, нами синтезированы 4 новых, ранее не описанных в литературе производных урацила, содержащих в положении N1 пиримидинового цикла ю -(3,5-диметилфенокси)алкильный фрагмент и различающихся длиной ациклической цепи, соединяющей ароматические ядра, изучены их спектральные и физико-химические свойства. Соединения этого ряда представляют большой интерес в качестве потенциальных противовирусных агентов.
