Расчет химических потенциалов компонентов наночастиц

Автор: Дрозин А.Д., Дудоров М.В., Куркина Е.Ю., Вяткин Г.П.
Журнал: Вестник Южно-Уральского государственного университета. Серия: Металлургия @vestnik-susu-metallurgy
Рубрика: Физическая химия и физика металлургических систем
Статья в выпуске: 2 т.24, 2024 года.
Бесплатный доступ
Получено простое выражение для расчета химических потенциалов компонентов в изотропных конденсированных наночастицах на основе соответствующих значений в объемных фазах. В первом приближении была учтена зависимость поверхностного натяжения от размера частиц. Для расчета необходимо знать зависимости от состава поверхностного натяжения и молярного объема соответствующей объемной фазы. Выражение удобно для изучения изменений состава наночастицы во время ее роста. Это выражение полезно для металлургии (образование неметаллических включений), аналитической химии (образование осадков), нанотехнологий (образование нанокристаллов в аморфных металлах) и других отраслей науки и техники. Предложенный метод может быть легко обобщен для анизотропных нанокристаллов.
Наночастицы, химический потенциал, поверхностные эффекты, термодинамика, неметаллические включения, рост наночастиц
Короткий адрес: https://sciup.org/147243664
IDR: 147243664 | УДК: 669.1 | DOI: 10.14529/met240201
Calculation of the chemical potentials of nanoparticles components
Simple and convenient expression for the calculation of the chemical potentials in isotropic condensed nanoparticles based on the corresponding value for the bulk phase has been obtained. In the first approximation, the dependence of surface tension on the particle size has been taken into account. For the recalculation, the surface tension and the molar volume of the corresponding bulk phase as a function of a composition should be set. Expression convenient to study composition changes during nanoparticle growth. The expression is useful for metallurgy (formation of nonmetallic inclusions), analytical chemistry (formation of precipitations), nanotechnology (formation of nanocrystals in amorphous metals), and other branches of science and technology. The proposed method can be easy generalized for anisotropic nanocrystals.
Текст научной статьи Расчет химических потенциалов компонентов наночастиц
Точное определение химических потенциалов компонентов наночастиц очень важно для нанотехнологий, особенно для изучения роста наночастиц сложного состава. Для расчета химического потенциала обычно применяется метод [1–5]. Энергия Гиббса GNano наночастицы определяется как
G
Nano
_ GBuUk + G$urface
где GBulk – энергия Гиббса объемной фазы с тем же составом и GSurface – дополнительная энергия Гиббса, связанная с поверхностным натяжением. Для расчета GBulk применяется метод CALPHAD с учетом дополнительного поверхностного давления. Стандартная энергия Гиббса наночастицы чистого i-го компо- нента определяется как
G0Nano _ G0 + CV i ir
где c i - коэффициент поверхностной энергии, Vi – молярный объем, r – радиус частицы, С – поправочный коэффициент, связанный с формой частицы и поверхностной деформацией.
В этой статье предлагается более общий и более удобный подход к расчету химических потенциалов.
Обычно выражение для химического потенциала i-го компонента системы записыва- ют в виде
^_И 0 (T, P) + RT In a., (3) где T – температура, P – давление, ai – активность i-го компонента и ц0 (T, P) - некоторое произвольно выбранное эталонное значение химического потенциала (стандартный химический потенциал). Обычно мы не знаем истинного значения ц0 (T, P), но это не ме- шает использовать его для изучения химических реакций. Если каждый компонент рассматривать при одинаковой температуре и давлении, то определенная комбинация членов ц0 (T, P) компонентов частицы равна RT lnK (T, P), где R - универсальная газовая постоянная и K – константа равновесия, которая известна для многих реакций в макросистемах.
Однако, если мы изучаем химические реакции в системе различных микро- и наночастиц, мы не можем поступить таким обра- зом. Из-за поверхностных эффектов давление в частице и некоторые другие свойства зависят от размера частицы. Мы не можем комбинировать члены ц0 (T, P) компонентов для получения RT ln K (T, P) и использования химических потенциалов для изучения химических реакций среди малых частиц. Это ограничивает наши возможности изучать образование и рост наночастиц во многих областях науки и техники. В этой статье мы выведем выражение для химического потенциала с общим стандартным условием для всех частиц.
Термодинамические соотношения
Рассмотрим наночастицу, состоящую из m компонентов, как термодинамическую систему при температуре T и давлении Pe , оказываемом окружающей средой на систему. Запишем для нее фундаментальное уравнение Гиббса в виде
TdS _ dU + PdV -c ds - ^ ц i dn i , (4)
i = 1
где S – энтропия, U – внутренняя энергия, P - давление в частице ( P ^ Pe ), V - объем, c - коэффициент поверхностного (или межфазного) натяжения, который, в общем случае, зависит от размера частицы, s – площадь поверхности частицы, m – количество компонентов и n i – количество молей i -го компонента.
Используя (4), мы можем записать полный дифференциал свободной энергии Гиббса
G _ U + PV - TS (5)
в виде dG _ -SdT + VdP + cds + ]m цdin.. (6)
i _ 1
Рассмотрим свободную энергию Гиббса как математическую функцию интенсивных переменных (T, P) и экстенсивных переменных (s, n1,...,nm). Химический потенциал цi зависит от этих же аргументов:
цi _цi (T,P, s, ni,..., nm), и его полный дифференциал может быть за- писан в виде d цРц dT + dP +
' dT dP m d^ +L ^d~dnj.
j ■ _i d nj
Так как из (6)
^ =4^1 дs дs l дni J
д дni
1^ 1 l дs J
ас д ni
и
= -S,, = V,
д T 1 дP
где Si и Vi – парциальная молярная энтропия и парциальный молярный объем i -го компо нента в системе. Используя (9) и (10), мы м ожем переписать (8) в виде
кой ( Ре , 0 ) . Зададим линию интегрирования в виде ломаной (см. рисунок) с вершинами B ( P ф , s ф) , C ( P ф ,0 ) , D ( Ре ,0 ) и получим
М D ) -н i ( B ) =
= J —(P, s) ds + V( P, s) dP
BCD i или
Mi( Pe ,0 )-н,( Pф, s ф) =
дс
d н i = VdP +—ds + (d н i )P ., д n- P,s
где ( d н i ) Ps — оставшаяся часть частных
дифференциалов d н i выражения (8), выраж ение ( d н i ) Ps равно 0 в процессах с постоя н-
ными T , n 1 , ..., nm .
Рассмотрим химический потенциал н i как математическую функцию интенсивных переменных ( T , P ) и экстенсивных переме нных ( s , n 1 ,..., n m ). Найдем приращение н i , когда давление P и площадь поверхности s изменяются от начального состояния ( P = P ф , s = s ф) к состоянию ( P = Ре , s = 0 ) ,
= J дС (Pф, s ) ds + J V( P, s ) dP ,
s ф ‘ni Pф ф e sф ‘С ф
н i =н i + ( P , s ) ds +
0 д n i
+ J P V i ( P ,0 ) dP . (12)
при котором постоянными.
н i ( p e ,0 ) =и е .
другие аргументы остаются Обозначим н i ( P ф , s ф) = н ф ,
Разность их равна кривол и-
нейному интегралу от d нi (P, s) вдоль линии фф с начальной точкой IP , s ) и конечной точ-
Зависимость поверхностного натяжения от размера частицы
Исследуем коэффициент поверхностного натяжения как математическую функцию интенсивных свойств ( T , P ) и экстенсивных свойств ( s , n 1 , ..., nm ) рассматриваемой системы. Поверхностное натяжение – интенсивное свойство системы; следовательно, с - однородная функция (степени 0) экстенсивных переменных s , n 1 ,..., nm , и мы можем записать [6]
с ( T , P , X s , X n i ,..., X n m ) =
= с ( T , P , s , n i ,..., nm ) , (13)
где X - произвольная постоянная. Положив
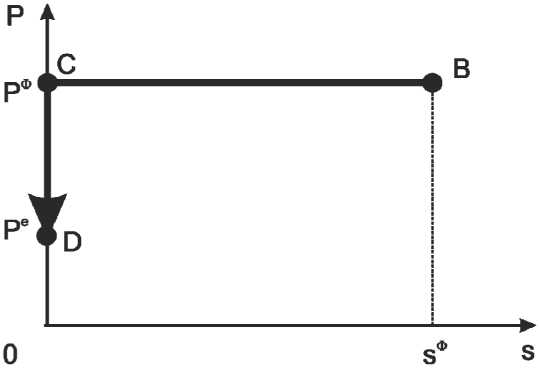
Линия интегрирования для криволинейного интеграла от дифференциала (11)
The path for line integral of the differential (11)
m
X = 1 n , где n = ^ n i - общее число молей в i = 1
частице, получаем
6 ( T , P , s , П 1 ,..., П т ) =
= 6 [ T , P , -, n 1 ,..., n m ] . (14)
к n n n J
Отметим, что ni- = Xi, (15)
n где Xi – мольная доля i-го компонента. Учтя, что
где 5 - расстояние между поверхностью натяжения и эквимолекулярной поверхностью, 6 - поверхностное натяжение для плоской поверхности (в отличие от 6). Приведем это выражение к соответствию с (17). Так как r = 3 nv/s, (22) из (21) получим
6=
s ,
1 + а
где а = — . Будем считать а постоянной ве-3 v
m
Z X i = 1,
i = 1
мы можем сделать вывод, что поверхностное натяжение может быть представлено в виде функции
6 = 6 I t , p , -, x 1 ,..., xm - 1 1 . (17) к n J
Чтобы избежать путаницы, сразу отметим, что для сферической частицы радиуса R мы имеем
V = 4 n r 3 , s = 4 n r 2 , (18)
P ф _ p^ = 22. (19) r
В таком случае P и s становятся взаимно зависимыми; 6 не остается однородной функцией переменных состояния, и использование теории однородных функций некорректно. Поэтому, если мы используем теорию однородных функций, мы должны рассматривать P и s как независимые переменные и
личиной. Заметим, что по определению 6 не может зависеть от кривизны. Поэтому мы можем использовать (17) для 6 в виде
6 = 6 ( T , P , X 1 ,..., X m - 1 ) . (24)
Теперь мы можем оценить первый интеграл в (12). Из (23) находим
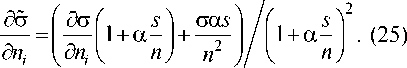
Принимая во внимание независимость 6
от s , получаем
г s фд<7/ ф \ n \ — I P , s ) ds =— J 0 5 ni ' а
, а 6
+ .
, s ф а
1 + а — n
г
учитывать различные зависимости только после получения конечных выражений.
Выражение (17) показывает, что площадь
поверхности s входит в наши уравнения только в составе комплекса s n . Заметим, что
выражение s n связано с кривизной поверх-
ности. В частности, для сферической частицы
s sv 3v n V r,
где v – средний молярный объем.
Найдем зависимость поверхностного на тяжения отs / n . Р.Ч. Толман [7] получил вы-
ражение
6 =
1 + 251
r
, ф А
n ^56 6 ,
- —+- | ln 1 + а— +
s
a^d n i
n
к
n J
s ф
Так как а— очень мало ( 5 « r ) , мы можем n
использовать приближения
ln 1 + а—
к
1 s ф
1 + а— n
n J
s ф а—, n
1 s ф
1 -а— n
и получить гsф д6 / ф \56
Jo Г(P ,s)ds = zs .
J 0 5ni v '5
В соответствии с (15), (24) имеем d6 = m-1 d6 dXj = m-1 d6 5 ij - Xj=
5n, дX, dn дX,n i J 1 j iJ i m-1
=-Z6 ":гX)
n J = 1
и
Ф . „Ф m -1
£ Н P * , * ) d S = “- '■<’ f X j ) (30)
0 d п , n ~ ~ 1y
, d' e [0, если i ^ j где 'j = и 5ij = 11 ■ • dXj [1, если i = j
5 -функ-
ция Кронекера.
Химический потенциал компонента
частицы
Вернемся к формуле (12). Рассмотрим второй интеграл. В первом приближении для конденсированной фазы можно предположить, что молярный объем не зависит от давления, тогда
PФ fpe Vi(P,0)dP = Vi(PФ-Pe). (31)
Используя (30) и (31), мы можем переписать (12) в виде
и : ,. e + v ( p ф - P ) +
Ф m - 1
+—E' ( 5 .j - j (32)
n j = 1
Теперь определим физический смысл члена p. e . Химический потенциал является интенсивным свойством системы, следовательно, он является однородной функцией (степени 0) экстенсивных переменных. Точно так же, как мы получили выражение (17) для ст ,
lim p Ф =и i ( Т , Pe ,0, X 1 ,..., X m - 1 ) . (36) n ^W
Сравнивая (35) и (36), можно заключить, что pe - это химический потенциал объемной фазы того же состава и температуры, что и наночастица, но под давлением Pe . Используя (3), мы можем записать и e = и 0 (Т, Pe) + RT ln a„ (37) где ai – активность i-го компонента и и0 (T, P) - некоторое произвольно выбранное
эталонное значение химического потенциала (стандартный химический потенциал).
Основная идея определения физического смысла члена иe основана на следующем
рассуждении: площадь поверхности s входит в выражение химического потенциала (33)
ss только как —. Если n ^ » , то--> 0 ; тот же nn
для химического потенциала можно получить
и i = и i I t , p ,-, x 1
£ n
., л m - 1
В частности,
ф и i
, Ф
= и i t , P Ф , —, X 1
£ n
,..
., X m - 1
J
.
Член pe в (12) был определен как химический потенциал в гипотетическом состоянии с фиксированными значениями P = Pe и - = 0. Следовательно, мы можем заключить, что иe =pi (Т,Pe,0,X1,...,Xm-1). (35) С другой стороны, пусть общее число молей в (33) стремится к бесконечности при постоянных температуре и составе. В этом случае частица становится обычной объемной фазой; дополнительное поверхностное давление исчезает, PФ становится равным давлению Pe, оказываемому окружающей средой. Соотношение s n будет стремиться к нулю. Мы получаем из (34)
результат мы получим, если предположить - = 0. Таким образом, гипотетическое состояние с - = 0 без физического смысла эквивалентно реальному термодинамическому состоянию объемной фазы того же состава и температуры.
Подставляя (37) в (32), получим и Ф = и 0 (Т, Pe) + V-( p Ф- Pe) +
Ф m - 1
+—E' j ( 5 ij - X j ) + RT ln a{ . (38)
n j = 1
Отметим, что в нашем рассмотрении мы учли зависимость поверхностного натяжения от размера частицы. Однако конечное выражение (38) содержит не реальное поверхностное натяжение ст , а поверхностное натяжение для объемной фазы ст.
Если частица имеет сферическую форму, используя (18) и (19), получим и,Ф=и0 (Т, Pe)+2V + m-1
+-E' j ( 5 j - X j ) + RT ln a i . (39)
r j = 1
Пример
В сталеплавильном производстве после расплавления шихты и окисления примесей расплав металла имеет избыток кислорода. Для удаления кислорода добавляют раскислители. Если используется алюминий, возможны следующие химические реакции:
2 [Al] + 3[O] = ( А120з ), [Fe]+[O]=(FeO)
и образуются наноразмерные зародыши продуктов реакций. Зародыши растут, и химические реакции (40) протекают на их поверхности. Растущие наночастицы состоят из раствора Al2O3-FeO. В окрестности поверхности частицы концентрации компонентов фазы расплава – Al, O, Fe – приходят в локальное равновесие с концентрациями компонентов фазы наночастиц – Al2O3, FeO .
Естественно, что концентрации компонентов фазы расплава в непосредственной близости от частицы не равны объемным концентрациям реагентов в расплаве. Это вызывает диффузионные потоки компонентов и доставку веществ для роста частицы.
Определим эти локальные равновесные концентрации реагентов на поверхности частиц разного размера и состава. Пусть температура расплава равна 1873 К. Воспользуемся следующими значениями [7]:
c ai2o3 = 1,40 н/м, ^ FeO = 0,18 н/м,
^Al2O3 - 3,433 -10 5 м3/моль, VFeO - 1,562 ’10—5 м3/моль, aAl2O3 23
K Al2O3 - 2 3 -5 10 , aAlaO
K FeO = " FeO = 440. a Fe a O
Обозначим долю оксида алюминия в
Запишем условия равновесия для химических реакций (40):
2 ^ [ Al ] + 3 ^ [ O ] =^ ( Al2O3 ) , ^ [ Fe ] + ^ [ O ] = ^ ( FeO ) .
Используя выражение (3) для химических по- тенциалов компонентов расплава, получаем
^ [ Al ]^ [Al ] ( T , P e ) + RT ln X [ Al ] ,
H [ O ] =H [O ] ( T , P e ) + RT In X [ O ] , ^ [ Fe ] =^ [Fe ] ( T , P e ) +
+ RT In ( 1 - X [ A ] - X [ o ] ) .
Воспользуемся выражением (38) для химических потенциалов компонентов частицы. Индекс 1 относится к Al 2 O 3 , индекс 2 относится к FeO . Получаем
» It !2\^'"V A ^
Ц ( Al2O 3 ) =^ ( Al2O 3 )( T , P ) + r +
+ —— a' ( x )( 1 - x ) + RT ln x ,
r
U 0 (T P2 2 °( x ) V ( Fe O )
^ ( FeO ) -^ ( FeO )( T , P ) + r
--— a' ( x ) x + RT ln ( 1 - x ) .
Подставляя (45), (46 ) в (44) и принимая во внимание, что
^ (Al2O3 ) - 2 ^ [Al ] - 3 ^ 0O ] - - RT ln K ( Al 2 O3 ) , ^ ( FeO ) - ^ (Fe ) - ^ 0O ] - - RT ln K ( FeO ) ’
частице через x ( x - Xai2o3 ) •
Считаем, что
получим систему уравнений:
зависимости поверхностного натяжения и молекулярного объема от состава частицы линейны:
X 2 X 3 1
Al O -------exp x K Al2O3
r 2 o V l2o3+MM 1
\
rRT
v - ( V Al 2 O3 V FeO ) x + V FeO -
( 1 - X Al
—
X oHo -
1 - x
= 1,871 - 10 - 5 x + 1,562 - 10 - 5 ( м3/моль ) ,
° = ( ° Al 2 O3 - ° FeO ) x + ° FeO = (42)
= 1,22 x + 0,18 ( Н/м ) , o'- 1,22 ( Н/м ) .
Для простоты предположим, что активности компонентов частицы и расплава равны их молярным долям:
a ( Al2O3 ) - x , a ( FeO ) = 1 x , a [ Al ] - X [ Al ] ,
a [ O ] - X [ O ] , a [ Fe ] = 1 - X [ Al ] - X [ O ] .
1 exp K FeO
(2o Vf„o - 3 v o' x/
[ RT )
.
Для примера, решая эту систему с раз-
личными значениями r и x , принимая во внимание (41), (42), мы рассчитали таблицу.
Таблица позволяет сделать некоторые выводы о природе роста частиц. Например, предположим, что расплав содержит 0,1 % алюминия и 0,05 % кислорода ( X A lk - 0,001, X Ouk - 0,0005). В соответствии с таблицей расплав вблизи поверхности зародыша с ра-
Локальные равновесные концентрации реагентов у поверхности частицы радиуса r и долей оксида алюминия x
Local equilibrium concentration near the surface of the particle with radius r and alumina fraction x
Заключение
Список литературы Расчет химических потенциалов компонентов наночастиц
- Park J., Lee J. Phase diagram reassessment of Ag-Au system including size effect // CALPHAD. 2008. Vol. 32, no. 1. P. 135-141. DOI: 10.1016/j.calphad.2007.07.004 EDN: YAGYEV
- Lee J., Park J., Tanaka T. Effects of interaction parameters and melting points of pure metals on the phase diagrams of the binary alloy nanoparticle systems: A classical approach based on the regular solution model // CALPHAD. 2009. Vol. 33, no. 2. P. 377-381. DOI: 10.1016/j.calphad.2008.11.001 EDN: YCGHUP
- Eichhammer Y., Heyns M., Moelans N. Calculation of phase equilibria for an alloy nanoparticle in contact with a solid nanowire // CALPHAD. 2011. Vol. 35, no. 2. P. 173-182. DOI: 10.1016/j.calphad.2011.02.002 EDN: WEHHMR
- Cu-Ni nanoalloy phase diagram - Prediction and experiment / J. Sopousek, J. Vrestal, J. Pinkas et al. // CALPHAD. 2014. Vol. 45, no. 2. P. 33-30. DOI: 10.1016/j.calphad.2013.11.004 EDN: YEWZOZ
- Lee J., Sim K.J. General equations of CALPHAD-type thermodynamic description for metallic nanoparticle systems // CALPHAD. 2014. Vol. 44. P. 129-132. DOI: 10.1016/j.calphad.2013.07.008 EDN: YEHEVJ
- Фихтенгольц Г.М. Курс дифференциального и интегрального вычисления. М.: Лань, 2024. 608 с.
- Кнюппель Г. Раскисление и вакуумная обработка стали: в 2 ч. М.: Металлургия, 1973. Ч. 1. 312 с.
