Роль гена PPM1D в канцерогенезе миелоидных неоплазий: молекулярные механизмы и перспективы таргетной терапии

Автор: Колосова Е.Д., Богданова Д.А., Демидов О.Н.
Журнал: Сибирский онкологический журнал @siboncoj
Рубрика: Обзоры
Статья в выпуске: 3 т.25, 2026 года.
Бесплатный доступ
Цель исследования обобщение накопленных знаний о роли фосфатазы PPM1D в клональном гемопоэзе (КГ) и патогенезе гематологических заболеваний, в частности при остром миелоидном лейкозе (ОМл), а также рассмотрение возможности терапевтического таргетирования PPM1D с помощью специфических ингибиторов. Материалы и методы. Поиск соответствующих источников проводился в базах данных Web of science, PubMed и scopus. Отбор публикаций осуществлялся на основании актуальности исследований и релевантности их тематики к теме обзора. Поиск научной литературы проводился с использованием следующих ключевых терминов: «mutations of PPM1D», «PPM1D and cancer», «clonal hematopoiesis», «PPM1D and clonal hematopoiesis», «PPM1D gene», «WiP1 phosphatase», «clonal hematopoiesis of indeterminate potential», «therapy-related AML», «p53 signaling pathway», «cell cycle regulation», «targeted cancer therapy», «hematologic malignancies», «truncating mutations» и «inhibitors of PPM1D». Было проанализировано 142 источника, из которых 63 были включены в обзор. Результаты. Клональный гемопоэз неопределенного потенциала (КГНП) характеризуется как состояние с соматическими мутациями в генах-драйверах миелоидных неоплазий при аллельной нагрузке >2 % в клетках крови у лиц без гематологических заболеваний. Распространенность КГНП превышает 10 % у лиц старше 60 лет и достигает 25 % у пациентов с онкологическими заболеваниями. Противоопухолевая терапия способствует селекции клонов гемопоэтических стволовых клеток с мутациями, в том числе с мутациями в гене PPM1D, что значительно повышает риск развития миелоидных новообразований, ассоциированных с предшествующей терапией (t-MN). Фосфатаза PPM1D является негативным регулятором p53 и многих путей клеточной гибели. Ее сверхэкспрессия детектируется при различных типах солидных опухолей (рак яичников, рак молочной железы и др.), а ее мутации обнаруживаются у 1-2 % пациентов с миелоидными неоплазиями de novo и у 10-20 % больных с t-MN. Последние исследования демонстрируют преобладающую роль мутаций PPM1D в возраст-ассоциированном клональном гемопоэзе, при укорочении теломер и наличии герминальных мутаций репарации ДНК, что подчеркивает ключевое значение этой фосфатазы в патогенезе миелоидных заболеваний. Заключение. PPM1D представляет собой перспективную терапевтическую мишень для лечения ОМЛ за счет своей ключевой роли в регуляции многих путей клеточной гибели и ответа на клеточный стресс. Ингибиторы PPM1D могут стать основой для разработки комбинированных схем терапии, особенно для пожилых пациентов и пациентов с приобретенной резистентностью к противоопухолевым препаратам.
PPM1D, клональный гемопоэз, острый миелоидный лейкоз, химиотерапия, ингибиторы PPM1D, таргетная терапия, клональный гемопоэз неопределенного потенциала
Короткий адрес: https://sciup.org/140315694
IDR: 140315694 | УДК: 616.155.392.8-08:615.277.3:577.21 | DOI: 10.21294/1814-4861-2026-25-3-96-109
The role of the PPM1D gene in the carcinogenesis of myeloid neoplasms: molecular mechanisms and prospects for targeted therapy
Objective: to summarize the accumulated knowledge on the role of PPM1D phosphatase in clonal hematopoiesis and the pathogenesis of hematological malignancies, particularly in acute myeloid leukemia, as well as to evaluate the feasibility of therapeutic targeting of PPM1D using specific inhibitors. Materials and Methods. a search for relevant sources was conducted in the Web of science, PubMed, and scopus databases. Publications were selected based on the relevance of the studies and the pertinence of their subject matter to the topic of the review, the literature search was performed using the following key terms: “mutations of PPM1D", “PPM1D and cancer”, “clonal hematopoiesis”, “PPM1D and clonal hematopoiesis”, “PPM1D gene”, “WIP1 phosphatase”, “clonal hematopoiesis of indeterminate potential”, “therapy-related AML”, “p53 signaling pathway”, “cell cycle regulation”, “targeted cancer therapy”, “hematologic malignancies”, “truncating mutations” and “inhibitors of PPM1D”. A total of 142 sources were analyzed, of which 63 were included in the review. Results. Clonal hematopoiesis of indeterminate potential (CHIP) is characterized as a condition involving somatic mutations in driver genes of myeloid neoplasms at a variant allele frequency of >2 % in blood cells of individuals without hematological disorders. The prevalence of CHIP exceeds 10 % in individuals over 60 years of age and reaches 25 % in cancer patients. Antineoplastic therapy promotes the selection of hematopoietic stem cell clones harboring mutations, including mutations in the PPM1D gene, which substantially increases the risk of developing therapy-related myeloid neoplasms (t-MN). PPM1D phosphatase is a negative regulator of p53 and numerous cell death pathways. Its overexpression is detected in various types of solid tumors (ovarian cancer, breast cancer, and others), while its mutations are identified in 1-2 % of patients with de novo myeloid neoplasms and in 10-20 % of patients with t-MN. Recent studies demonstrate the predominant role of PPM1D mutations in age-associated clonal hematopoiesis, in the context of telomere shortening, and in the presence of germline DNA repair mutations, thereby underscoring the pivotal significance of this phosphatase in the pathogenesis of myeloid disorders. Conclusion. PPM1D represents a promising therapeutic target for the treatment of AML owing to its key role in the regulation of multiple cell death pathways and the cellular stress response. PPM1D inhibitors may serve as a foundation for the development of combination therapy regimens, particularly for elderly patients and those with acquired resistance to antineoplastic agents.
Текст научной статьи Роль гена PPM1D в канцерогенезе миелоидных неоплазий: молекулярные механизмы и перспективы таргетной терапии
Клональный гемопоэз (КГ) представляет собой экспансию клеток-потомков одной гемопоэтической стволовой клетки (ГСК), которая приобрела соматические мутации [1]. Одним из вариантов КГ является клональный гемопоэз неопределенного потенциала (КГНП). Клональный гемопоэз неопределенного потенциала определяется как состояние с соматическими мутациями в различных генах, в том числе и генах-драйверах миелоидных неоплазий при аллельной нагрузке ≥2 % в клетках крови у лиц без гематологических заболеваний [2]. Частота КГНП увеличивается с возрастом, а его распространенность среди лиц старше 60 лет составляет более 10 % [2, 3]. Воздействие химиотерапии и лучевой терапии способствует развитию КГНП, особенно в клонах с мутациями в генах, участвующих в реакции на повреждение ДНК (DDR), таких как TP53, PPM1D, ATM или CHEK2 [4]. КГНП может предшествовать миелоидным злокачественным новообразованиям и наблюдается примерно у 25 % пациентов с опухолями различного генеза [4]. Несмотря на то, что у подобной непатологической экспансии клонов неопределенный потенциал, КГНП является фактором риска развития миело-диспластических синдромов (МДС) или острого миелоидного лейкоза (ОМЛ) [4].
PPM1D (протеинфосфатаза Mn2+/Mg2+-зависи-мая 1D) принимает участие в сигнальном пути DDR, являясь негативным регулятором p53 – ключевого медиатора апоптотических процессов, – а также дефосфорилирует ATM, ATR, CHK1 и CHK2 [5–7]. Влияя на фосфорилирование p53, PPM1D влияет на баланс между выживаемостью клетки и ее гибелью путем апоптоза. Фосфатаза PPM1D также регулирует другие каскады клеточной гибели, такие как ферраптоз, сенесценция и аутофагия [7, 8]. PPM1D сверхэкспрессируется вследствие стабилизирующих мутаций или амплификаций при различных первичных опухолях человека, а мутации PPM1D наблюдаются у 1–2 % пациентов с миелоидными новообразованиями de novo, но их частота в значительной степени увеличивается при t-MN (10–20 %) [9, 10]. Последние исследования указывают на преобладающую роль мутаций PPM1D в КГ, ассоциированном с возрастом и укорочением теломер, а также при наличии герминальных мутаций в генах системы репарации ДНК [11–14]. Мутации PPM1D также являются одними из наиболее частых событий при хроническом лейкозе, связанном с терапией [15–19].
В данном обзоре мы суммируем накопившиеся знания о роли PPM1D в КГ и миелоидных неоплазиях, в частности при ОМЛ. Мы анализируем не только роль данной фосфатазы в патогенезе гематологических заболеваний, но и рассматриваем возможность терапевтического таргетирования PPM1D.
Структура фосфатазы PPM1D
PPM1D относится к семейству протеинфосфа-таз 2C-типа (PP2C) и представляет собой фермент, который у человека кодируется геном PPM1D [5–7]. Белкам этого большого семейства, в том числе PPM1D, свойственно наличие каталитических и регуляторных доменов. Каталитические домены являются крайне консервативными, в то время как регуляторные домены значительно варьируют, обеспечивая специфичность субстратов и функциональное разнообразие этих белков [5, 6].
От каталитического домена белка PPM1D отходят 3 петлевые структуры: А-петля, B-петля и C-петля, из которых В-петля характеризуется наличием сигнала ядерной локализации [6]. В контексте роли B-петли в детерминации субстратной специфичности установлено, что кислотные аминокислотные остатки, локализованные в непосредственной близости к сайтам фосфорилирования ключевых субстратов (ATM и p53), выполняют критическую функцию в процессе их дефосфорилирования фосфатазой PPM1D [6, 20]. Эта уникальная характеристика B-петли дает возможность разрабатывать ингибиторы, которые являются высокоспецифичными для PPM1D. Таким высокоспецифичным ингибитором PPM1D является GSK2830371 [6, 21].
Мутации гена PPM1D
Ген PPM1D локализован на длинном плече 17-й хромосомы (17q) и состоит из 7 экзонов. Благодаря альтернативному сплайсингу формируются различные изоформы мРНК, содержащие 6 или 7 экзонов [5, 14]. Геномные аберрации PPM1D при различных типах рака (рак яичников, рак молочной железы и др.) могут проявляться в виде амплификации гена [14]. Амплификация приводит к увеличению копий гена и сверхэкспрессии транскрипта PPM1D, что также приводит к повышению активности фосфатазы и нарушению сигнализации DDR [14]. В солидных опухолях амплификации PPM1D и мутации TP53 часто взаимоисключающие, что указывает на их общую роль в инактивации контрольной точки p53 [14]. Также на хромосоме 17q расположено несколько других генов, ассоциированных с развитием рака (BRCA1, HER2 и др.). Следовательно, сверхэкспрессия PPM1D при амплификациях 17q может действовать совместно с повышенной экспрессией других онкогенов, способствуя онкогенезу [14].
Большинство мутаций в гене PPM1D приводят к потере С-концевой части белка, ответственной за протеасомную деградацию, что приводит к стабилизации и увеличению количества белка, обладающего полноценной каталитической активностью (рис. 1) [22]. Укороченный белок PPM1D может накапливаться в клетке в количестве, в 16 раз превышающем количество полноразмерного PPM1D, даже в отсутствие клеточного стресса [14, 22]. Эти мутации приводят к тому, что повышенные уровни PPM1D могут эффективно подавлять каскады сигнализации, вызванные DDR, которые обычно опосредуются p53, ATM, CHK1 и CHK2 [14].
Наиболее изученной является мутация c.1654C>T (p.R552*), приводящая к образованию преждевременного стоп-кодона, которая часто выявляется в клетках костного мозга (КМ) у пациентов с различными злокачественными новообразованиями [6]. Мутации со сдвигом рамки считывания, которые приводят к D470fs или A481fs, и другие нонсенс-мутации, такие как Q510* и R552*, часто коррелируют с предшествующим воздействием химиотерапии или лучевой терапии [19]. Эти мутации приводят к гиперактивности белка PPM1D, который постоянно дезактивирует пути DDR, тем самым способствуя неконтролируемой пролиферации клеток, нарушению апоптоза и устойчивости к химиопрепаратам [16].
В нормальных условиях после повреждения ДНК, если оно необратимо, клетки быстро активируют p53, что приводит к остановке клеточного цикла и запуску процессов репарации или апоптоза [5, 23]. Аномально высокие уровни PPM1D, возникающие в результате амплификации или мутаций усечения гена, нарушают активацию p53, ослабляя критические контрольные точки клеточного цикла, такие как переход G1/S и контрольная точка G2/M, что в конечном итоге обеспечивает выживание и пролиферацию клетки с повреждениями ДНК [14, 22].
В настоящее время обнаружено, что PPM1D амплифицируется и сверхэкспрессируется в различных опухолях [14]. Мутации PPM1D в клетках
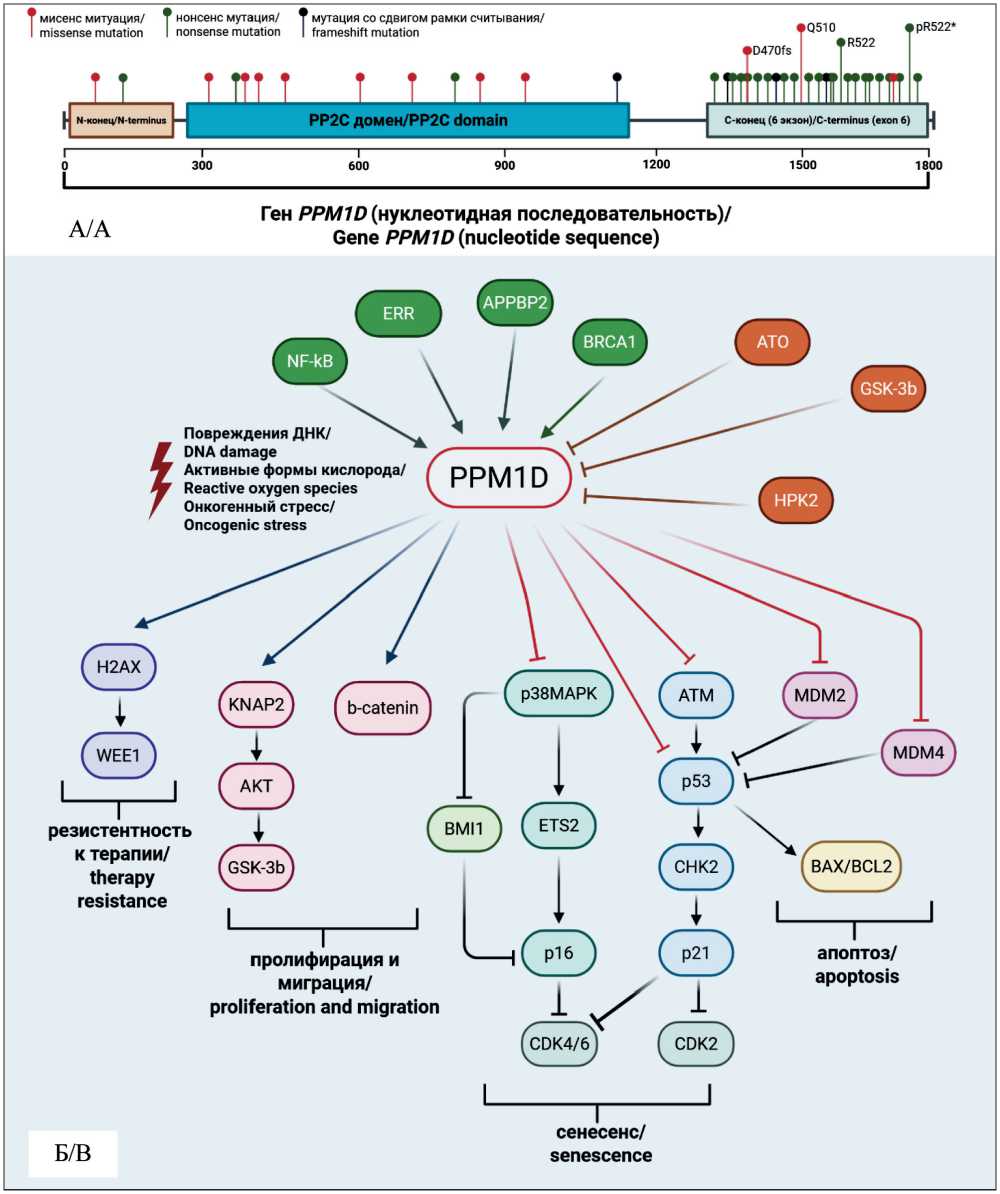
Рис. 1. А – схема структуры гена PPM1D , локализация и типы мутаций; Б – мишени фосфатазы PPM1D и функциональные последствия сигнальных путей, связанных с PPM1D.
Примечание: created in BioRender. Pukhalskaya, T. (2025)
Fig. 1. A – schematic representation of the PPM1D gene structure, illustrating the localization and types of identified mutations;
B – substrates of PPM1D phosphatase and the functional consequences for PPM1D-associated signaling pathways.
Note: created in BioRender. Pukhalskaya, T. (2025)
кроветворной системы ассоциированы с повышенным риском развития как солидных опухолей, так и злокачественных новообразований системы крови [14]. Кроме того, мутации в PPM1D при КГ приводят к клональному росту и к последующему увеличению риска возникновения злокачественных опухолей [14]. При гематологических патологиях, таких как t-MN и МДС, мутации PPM1D часто возникают вследствие предшествующего воздействия цитотоксических препаратов и связаны с клональной экспансией ГСК, несущих мутации PPM1D [5]. Аналогичным образом в солидных опухолях либо амплификации, либо мутации гена, связанные с укороченным вариантом PPM1D, ассоциированы с агрессивным фенотипам заболевания, устойчивости к химиотерапии и лучевой терапии, а также, в целом, неблагоприятным клиническим исходам [14, 23, 24].
Сигнальные пути – мишени PPM1D
При клеточном стрессе, повреждении ДНК или активации онкогенов (онкогенном стрессе) киназы ATM, ATR, CHK1 и CHK2 фосфорилируют p53 преимущественно по остаткам Ser15, Ser20, Ser33, Ser37 и Ser46 в N-концевом домене [25–28]. Эти модификации стабилизируют белок p53 и предотвращают его взаимодействие с негативным регулятором MDM2 [25]. Это фосфорилирование необходимо для накопления p53 и запуска транскрипции генов, участвующих в остановке клеточного цикла, апоптозе, сенесценции и репарации ДНК [26, 27].
После завершения процесса репарации ДНК фосфатазы обеспечивают отрицательную обратную связь, инактивируя компоненты DDR. Так, PPM1D индуцируется р53-зависимым образом и дефосфорилирует p53 по остаткам Ser15, Ser33 и Ser46, тем самым восстанавливая восприимчивость p53 к убиквитинированию, опосредованному MDM2, и последующей деградации, позволяя клетке избежать клеточной гибели и закончить репарацию ДНК [26–28]. Эта обратная связь крайне важна для поддержания клеточного гомеостаза и предотвращения негативных последствий, связанных с постоянной активацией p53, таких как преждевременная гибель клеток [7]. Апоптоз, регулируемый p53, широко изучался как ключевой процесс, сопровождающий лечение опухолей [29]. Дефосфорилирование p53 снижает транскрипцию проапоптотических генов, таких как BAX , PUMA и NOXA , тем самым предотвращая апоптоз при достаточной репарации DDR [7, 29]. В опухолях, содержащих дикий тип p53, аномальная экспрессия или повышенная активность фосфатаз, таких как PPM1D, связана с ослаблением апоптотического пути клеточной гибели, что приводит к выживанию опухолевых клеток и развитию химиорезистентности [7, 28]. PPM1D также напрямую инактивирует ось p38-MAPK путем дефосфорилирования сайта активации Thr180 и Tyr182 в условиях стресса, таких как воздействие УФ-излучения или H2O2 [30]. р38 может активироваться одновременно с р53 и оказывать позитивную посттрансляционную регуляцию р53 [30]. Несмотря на возможные перекресты между путями MAPK и p53, важно различать p53-зависимые и p38-зависимые эффекты делеции и/или активации PPM1D [28, 29, 31].
Сенесценция (клеточное старение) – состояние постоянной остановки клеточного цикла, которое является еще одним результатом активации сигнального пути p53. PPM1D модулирует сигнальный путь p53–p21, критически важный для индукции клеточного старения. Дефосфорилируя p53, эта фосфатаза снижает его способность индуцировать p21, тем самым предотвращая переход клеток в сенесцентное состояние [29]. Напротив, дефицит PPM1D приводит к повышению активности p53 и преждевременной сенесценции, о чем свидетель- ствуют повышенное фосфорилирование γ-H2AX и повышенный уровень p21 [14]. Регулирование данного каскада имеет решающее значение не только при нормальном старении клеток, но и в контексте рака, где уклонение от клеточного старения является отличительным признаком прогрессирования опухоли [32]. В литературе имеются также данные, что PPM1D может участвовать и в других типах клеточной гибели, в том числе в регуляции ферроптоза посредством влияния на экспрессию SLC7A11, аутофагии и др. [14].
Контролирование ответа р53 жизненно важно для предотвращения гибели или старения клеток, этот процесс особенно важен для гомеостаза стволовых клеток. Ингибирование PPM1D может повысить чувствительность р53 к восходящей передаче сигналов и таким образом предотвращать онкогенную трансформацию. Это показано у мышей с генетической делецией в гене Ppm1d, у которых путь р53 является активным [33]. В этой модели опосредующая роль p53 была подтверждена путем обращения вспять опухолесупрессивного фенотипа мышей Ppm1d–/–путем делеции Trp53 [33]. В то же время делеция Ppm1d не предотвращает лимфомы и саркомы, связанные с генотипом Trp53–/– [34]. Негативные последствия делеции Ppm1d, такие как преждевременное старение, лимфопения, также могут быть связаны с аномальной активацией пути р53 и во многих случаях предотвращаются ингибированием р53. Обнаружено, что делеция Ppm1d защищает мышей от онкогенеза молочной железы. При этом другие варианты мутаций в гене PPM1D (сдвиг рамки считывания и образование преждевременного стоп-кодона) приводят к синтезу укороченного и более стабильного белка, который сохраняет каталитическую активность и не подвергается нормальной деградации, тем самым фенокопируя сверхэкспрессию PPM1D. Такие мутации ассоциированы с развитием рака молочной железы, яичников и глиомы [34]. PPM1D дефосфорилирует H2AX по остатку Ser139, что приводит к снижению привлечения белков репарации, таких как MDC1, 53BP1 и BRCA1 [14, 23]. Удаляя фосфорилированные метки H2AX, PPM1D предотвращает длительную остановку контрольных точек и апоптоз клетки, что в опухолевых клетках может способствовать развитию резистентности к противоопухолевой терапии [14, 22]. Через инактивацию CHK1 PPM1D снижает активацию киназы WEE1, которая в норме фосфорилирует и инактивирует CDK1 [35]. В результате происходит преждевременная активация CDK1, что приводит к пролиферации с нерепарирован-ной ДНК [35]. Также PPM1D может влиять на сигнальные пути бета-катенина за счет регуляции ASPP2, негативного регулятора ядерной транслокации бета-катенина. Сверхэкспрессия PPM1D приводит к снижению экспрессии ASPP2, что, в свою очередь, разрушает тройной комплекс ASPP2- бета-катенин-E-кадгерин, затем, попав в ядро, бета-катенин активирует целевые гены, управляющие прогрессированием клеточного цикла и пролиферацией (рис. 1) [35].
Роль PPM1D в КГ
ГСК дают начало всем клеткам крови и обладают уникальными морфологическими и функциональными характеристиками, которые могут нарушаться при развитии патологических процессов [11]. Функционирование и самообновление ГСК строго регулируются сложной сетью супрессоров опухолей, включая p53, ATM и INK4a/ p16 [11]. В последние годы активно исследуется феномен КГ, при котором происходит клональная экспансия клеток крови, несущих соматические генные аберрации [24, 36–39]. В современной терминологии КГ используется для обозначения любой клональной экспансии, в то время как КГНП описывает клональные мутации в генах, ассоциированных с лейкемией, при частоте аллельных вариантов ≥2 % [5, 40, 41]. Масштабные исследования секвенирования целого экзома образцов красного КМ в 2014 г. показали увеличение частоты КГ, зависимое от возраста, с распространенностью примерно 10 % среди лиц старше 60 лет [42]. Хотя ранние исследования указывали на то, что КГ является исключительно следствием стохастических событий, связанных со старением, недавние исследования показали функциональную роль конкретных генов в обеспечении усиленного самообновления, нарушения дифференцировки и устойчивости к стрессу, вызываемому внешними стимулами [1, 18, 24, 38]. В частности, эпигенетические регуляторы, такие как DNMT3A , TET2 и ASXL1 , оказались доминирующими драйверами, учитывая высокую частоту мутирования этих генов (таблица) [1, 18, 37, 38].
В норме эти гены регулируют метилирование ДНК и состояние хроматина, а наличие мутаций в данных генах часто приводит к изменению профиля экспрессии генов в общем, уклонению от сигналов дифференцировки и аномальным реакциям на воспалительные стимулы [38]. Мутации в факторах, традиционно связанных с онкогенной сигнализацией и ответом на DDR, например JAK2 и TP53 , встречаются с субклинической частотой, но они очень важны при КГ, связанном с терапией, или у лиц с дополнительными стрессорными факторами [14, 36, 39, 43]. Как показали полногеномные исследования, мутации в генах TERT , CD164 , TCL1A , TCL1B , SLC12A7 и PARP1 выявляют значительно реже [18, 24, 38, 40]. Хотя эти гены мутируют с меньшей частотой по сравнению с DNMT3A , TET2 или ASXL1 , их мутации стабильно детектируются в исследованиях, использующих расширенные панели генов и секвенирование, и могут влиять на поддержание теломер, клеточную адгезию и репарацию ДНК [22, 36, 39].
Особое значение в развитии КГ имеют укороченные мутации PPM1D [37, 44]. Эти мутации приводят к экспрессии стабильного и укороченного белка, который сохраняет ферментативную активность и позволяет ГСК избегать клеточной гибели, вызванной химиотерапией [37, 44]. Соматические мозаичные мутации PPM1D также обнаруживаются в периферической крови здоровых людей с возрастом и связаны с развитием КГ [45]. КГ, возникающий из мутировавших ГСК с селективным преимуществом, в настоящее время признан фактором риска развития гематологических злокачественных новообразований и сердечно-сосудистых заболеваний [45]. Мутации в гене PPM1D являются одним из ключевых драйверов КГ, что подчеркивает важность нарушений нормальной регуляции и функционирования ГСК в патогенезе КГ и последующей малигнизации кроветворной системы. Укорачивающие мутации в терминальном экзоне PPM1D придают гемопоэтическим клеткам химиорезистентность, обеспечивая их селективное размножение при воздействии химиотерапии, что объясняет высокую частоту этих мутаций у пациентов, получавших цитостатическое лечение по поводу наличия солидных опухолей [9, 10, 15, 16, 44]. Исследования, анализирующие частоту аллельных вариантов, показали, что даже редкие по частоте мутации PPM1D , часто обнаруживаемые у пациентов с клональными цитопениями или КГ, связанным с терапией, часто занимают доминирующие позиции в кроветворном компартменте, что указывает на их функциональную роль в клональной эволюции [16]. Клинические данные свидетельствуют о том, что у пациентов, получающих терапию на основе платины или радионуклидов, с большей вероятностью выявляются доминирующие мутации PPM1D , которые связаны с последующими нежелательными явлениями, такими как развитие миелоидных новообразований, связанных с терапией, включая t-AML [14]. Кроме того, интервал между цитотоксической терапией и обнаружением мутаций PPM1D может быть коротким, что позволяет предположить, что клоны с мутацией PPM1D могут существовать изначально с низкой частотой и быстро размножаться при воздействии генотоксического стресса, тем самым ускоряя развитие лейкемии [35, 45]. Недавнее полногеномное секвенирование клеток красного костного мозга 647 пациентов с мутацией PPM1D при КГ показало, что наблюдаемые изменения, по-видимому, обусловлены не только перенесенной химиотерапией, как считалось ранее, но и в большей степени процессами репликативного старения, сопровождающимися укорочением теломер [46].
Одним из важных открытий последних исследований является двойственная роль PPM1D в регуляции как самообновления, так и дифференцировки ГСК [11]. С одной стороны, повышенная активность PPM1D в ГСК приводит к нарушению
|
Таблица/table Основные драйверные мутации при КГ и доля КГНП Principal driver mutations associated with clonal hematopoiesis and the percentage frequency of their oc- |
|||
|
currence i |
n clonal hematopoiesis of indeterminate potential |
||
|
Ген/ Gene |
Функция/ Function |
Эффекты/механизмы мутаций/ Effects/mechanisms of mutations |
Доля КГНП/ Percentage of clonal hematopoiesis of indeterminate potential cases |
|
DNMT3A |
Метилирование de novo / de novo DNA methylation |
R882 и другие варианты мутаций действуют доминантно-негативно, что приводит к гипометилированию, нарушению дифференцировки и усиленному самообновлению ГСК [36]/ The R882 hotspot and other variants act in a dominantnegative manner, causing global hypomethylation, impaired differentiation, and enhanced self-renewal of HSCs [36] |
~10–60 % [24, 36–39] |
|
TET2 |
Фермент, опосредующий деметилирование ДНК/ Enzyme that mediates DNA demethylation |
Мутации с потерей функции приводят к снижению превращения 5-mC в 5-hmC, окислению, влияющему на активное деметилирование ДНК, приводящее к аберрантному гиперметилированию и нарушению регуляции генов [43]/ Loss-of-function mutations reduce the conversion of 5-mC to 5-hmC and its further oxidation, hindering active DNA demethylation, which results in aberrant hypermethylation and dysregulated gene expression [43] |
~5–20 % [24, 36, 38, 39] |
|
ASXL1 |
Фактор ремоделирования хроматина/ Chromatin-remodeling factor |
Укороченные мутации нарушают функцию поликомбного репрессивного комплекса, изменяя модификации гистонов [43]/ Truncating mutations disrupt Polycomb repressive-complex function, altering histone modifications [43] |
~7–30 % [24, 36–38] |
|
JAK2 |
Нерецепторная тирозинкиназа/ Non-receptor tyrosine kinase |
Мутация V617F приводит к конститутивной активации сигнальных путей [36, 39]/ The V617F mutation leads to constitutive activation of downstream signalling pathways [36, 39] |
~3–3,7 % [36, 39] |
|
TP53 |
Опухолевый супрессор, регулирующий клеточный цикл, апоптоз и др./ Tumor suppressor regulating cell cycle, apoptosis, etc. |
Мутации нарушают реакцию на повреждение ДНК и апоптоз, что приводит к геномной нестабильности [39]/ Mutations impair the DNA-damage response and apoptosis, leading to genomic instability [39] |
~1,9 % [39] |
|
PPM1D |
Фосфатаза, участвующая в реакции на повреждение ДНК/ Phosphatase involved in the DNA-damage response |
Укороченные мутации обеспечивают химиорезистентность и позволяют клонам HSC выживать [36, 39]/ Truncating mutations confer chemoresistance and allow HSC clones to survive [36, 39] |
~1,6–3,8 % [36, 39] |
Примечание: таблица составлена авторами.
Note: created by the authors.
апоптоза и усилению пролиферации в ответ на воздействие агентов, повреждающих ДНК, что способствует клональной экспансии [14, 40]. С другой стороны, укороченный белок PPM1D может влиять на баланс между миелоидной и лимфоидной дифференцировкой [47]. Например, повышенная активность белка PPM1D связана со смещением в гемопоэтической дифференцировке, при котором предпочтение отдается миелоидным клеткам, что может способствовать развитию предлейкозного состояния, наблюдаемого при миелоидных новообразованиях, связанных с терапией [47]. Более того, изменения в сигнальных путях, таких как mTOR и NOTCH, связаны с уменьшением способности к самообновлению и изменением клеток в мутантных ГСК, тем самым еще больше способствуя возникновению ОМЛ [11, 47].
Еще одним интересным направлением исследований является взаимодействие мутаций PPM1D с другими генетическими изменениями, в первую очередь с мутациями в гене TP53. Хотя мутации TP53 давно известны как факторы КГ и неблагоприятных исходов при ОМЛ, ряд исследований показывает, что мутации PPM1D могут возникать как независимо, так и в сочетании с мутациями TP53 [17, 38, 45]. Некоторые данные свидетельствуют о том, что, когда мутации PPM1D возникают при отсутствии изменений в гене TP53, они могут обусловливать меньшую геномную нестабильность [12, 14].
Роль PPM1Dв миелопролиферативных неоплазиях
Миелоидные неоплазии – злокачественные новообразования системы крови, возникающие из клеток миелоидного ростка кроветворения. К ним относятся ОМЛ, МДС, хронический миелоидный лейкоз и хронический миеломоноцитарный лейкоз [36]. Учитывая высокую частоту трансформации хронических миелоидных неоплазий и МДС в острые лейкозы (ОЛ), особую актуальность для изучения представляет ОМЛ, как наиболее распространенная и прогностически неблагоприятная форма миелоидных новообразований [36]. ОЛ прежде всего характеризуются неконтролируемой пролиферацией, нарушением дифференцировки и накоплением в КМ и периферической крови незрелых гемопоэтических клеток [36]. Эти злокачественные клетки постепенно замещают и ингибируют рост и созревание нормальных гемопоэтических предшественников и инфильтрируют различные органы и ткани [1, 36, 37]. Остаточная способность к дифференцировке лежит в основе фенотипической классификации заболевания [36, 38]. Известно, что это клинически гетерогенное заболевание со значительной вариабельностью выживаемости после лечения, на которую влияют такие факторы, как возраст, морфология бластных клеток, цитогенетические аномалии и генные мутации [38].
Клеточные клоны с мутациями гена PPM1D не только демонстрируют повышенную устойчивость к цитотоксическому стрессу, но и могут влиять на микроокружение КМ посредством изменения секреции цитокинов и провоспалительной сигнализации [39]. Экспериментальные данные на мышах, клетки которых экспрессируют укороченный белок PPM1D, показывают, что экспрессия мутантного гена PPM1D связана с усилением воспалительных реакций и даже сердечно-сосудистыми осложнениями, такими как неишемическая сердечная недостаточность, что позволяет предположить, что вследствие мутаций PPM1D могут происходить плейотропные эффекты, выходящие за рамки гемопоэтического компартмента [39]. Это системное воздействие может дополнительно способствовать развитию лейкоза путем развития хронического воспаления [40, 41]. Экспериментальные данные, полученные на мышах, экспрессирующих укороченный белок PPM1D, показывают, что, хотя мутация подавляет самообновление ГСК в условиях покоя, она значительно способствует развитию агрессивного ОМЛ в сочетании с генотоксическим стрессом [42]. Кроме того, анализ экспрессии генов в этих моделях выявил транскриптомные сигнатуры, которые коррелируют с неблагоприят- ным прогнозом при ОМЛ, что еще раз подчеркивает биологическую значимость мутантного гена PPM1D в развитии заболевания [43].
Интересным аспектом мутаций PPM1D является их мозаичная природа, особенно при гематологических злокачественных новообразованиях и в контексте КГ [44]. Мозаичные мутации PPM1D присутствуют только в части клеток и обнаруживаются при относительно низкой аллельной нагрузке. Они дают клеткам преимущество роста при генотоксическом стрессе [44]. Это явление особенно заметно на t-MN и МДС, когда после химиотерапии происходит экспансия мутантного клона, что способствует прогрессированию заболевания [5, 45].
Современная терапия ОМЛи стратегия таргетирования PPM1D
Понимание того, что ОМЛ является гетерогенным заболеванием с разнообразными генетическими, эпигенетическими и иммунологическими отклонениями, способствовало развитию стратегий лечения, основанного на объединении традиционной химиотерапии с таргетными препаратами, иммунотерапией и персонализированными режимами поддерживающей терапии, с мониторингом минимальной остаточной болезни (МОБ) [48].
Острый миелоидный лейкоз исторически лечили интенсивной химиотерапией, такой как схема «7 + 3» с цитарабином и антрациклином, а также консолидирующей трансплантацией аллогенных гемопоэтических стволовых клеток у пациентов с благоприятным соматическим статусом. Однако несмотря на десятилетия использования, результаты традиционной химиотерапии остаются неоптимальными, особенно у пожилых пациентов или у лиц с тяжелыми сопутствующими заболеваниями, а частота рецидивов остается высокой [48]. Достижения в области геномного профилирования не только улучшили прогнозирование за счет идентификации мутаций, ассоциированных с рецидивами, таких как FLT3 , IDH1/2 , NPM1 и TP53 , но и проложили путь к разработке таргетных препаратов, которые напрямую воздействуют на эти молекулярные сигнатуры [48].
Одним из первых прорывов в таргетной терапии стало использование ингибиторов FLT3. Мутации FLT3, которые встречаются примерно в 30 % случаев ОМЛ, ассоциированы с аномальной пролиферацией клеток, а ингибиторы, такие как мидостаурин, продемонстрировали преимущества в плане выживаемости при добавлении к стандартной химиотерапии у впервые диагностированных пациентов [49–51]. В условиях рецидива или рефрактерности новые ингибиторы FLT3, такие как гилтеритиниб, продемонстрировали улучшение показателей эффективности и выживаемости [52, 53]. Аналогичным образом были разработаны низкомолекулярные ингибиторы, направленные против мутантных форм IDH1 и IDH2, а именно ивосидениб и энасидениб соответственно, для противодействия онкогенным эффектам, возникающим из-за продукции 2-гидроксиглутарата и других соединений, которые вызывают гиперметилирование ДНК и блокируют дифференцировку лейкозных бластов [53, 54].
Для пожилых пациентов или пациентов с серьезными сопутствующими заболеваниями схемы низкой интенсивности становятся все более важными. Гипометилирующие агенты, такие как азацитидин и децитабин, уже давно используются у таких пациентов [54]. Одним из важных этапов в лечении данной когорты пациентов стало использование венетоклакса, селективного ингибитора BCL2, в сочетании с гипометилирующими агентами или низкодозным цитарабином [45, 55]. Эта комбинация улучшила ответ на терапию у пациентов и повысила уровень общей выживаемости у пожилых пациентов с ОМЛ, ранее не получавших лечения, продемонстрировав, что таргетирование антиапоптотических путей может преодолеть резистентность бластных клеток к классическим схемам цитотоксической терапии [54, 55]. Успех этих схем знаменует собой сдвиг в сторону использования комбинаций таргетных препаратов с низкоинтенсивной химиотерапией, обеспечивая эффективные варианты лечения [52]. В этой связи активное внимание исследователей направлено на изучение комбинированных схем на основе вене-токлакса в доклинических моделях [48, 54].
Помимо таргетной терапии, иммунотерапевтические стратегии стали перспективным направлением в лечении ОМЛ [48, 54]. Активно изучаются моноклональные антитела, конъюгаты антител с лекарственными препаратами, биспецифические активаторы Т-клеток и Т-клетки, несущие химерный антигенный рецептор, на ранней фазе терапии [48, 54]. Например, гемтузумаб озогами-цин, конъюгат антитела к CD33 с лекарственными препаратами, был повторно одобрен для лечения CD33-положительного ОМЛ после первоначальной отмены из-за опасений по поводу токсичности [48, 54]. Другие иммунотерапевтические подходы включают воздействие на иммунные контрольные точки, такие как PD1 и CTLA4, для усиления активации Т-клеток на антигенный стимул, а также новые агенты, такие как магролимаб, который воздействует на рецептор CD47, экспрессируемый на лейкемических клетках [54].
Значительным достижением в лечении миелоидных новообразований является интеграция персонализированной медицины посредством комплексного молекулярного профилирования и мониторинга МОБ. Технологии секвенирования нового поколения позволяют выявить мутации, требующие вмешательства в большинстве случаев ОМЛ, что дает возможность врачам адаптировать терапию к специфическим молекулярным и генетическим профилям каждого пациента [51, 56]. При ОМЛ в образцах пациентов и клеточных линиях обнаруживаются мутации, связанные с экспрессией укороченной формы белка PPM1D, эти изменения коррелируют с характеристиками заболевания высокого риска, плохой стратификацией риска и устойчивостью к классическим схемам химиотерапии [14]. Эксперименты по подавлению активности PPM1D на клеточных линиях ОМЛ продемонстрировали, что истощение PPM1D приводит к выраженной остановке клеточного цикла, снижению пролиферации и усилению апоптоза посредством активации сигнальных путей p38MAPK и p53 [57]. Для фармакологического воспроизведения этих эффектов разработан аллостерический ингибитор GSK2830371, селективно воздействующий на PPM1D и восстанавливающий фосфорилирование p53 и Chk2, тем самым реактивируя DDR в клетках ОМЛ [20]. Примечательно, что механизм действия ингибиторов PPM1D не ограничивается исключительно p53-зависимыми путями. Показано, что подавление PPM1D способно оказывать терапевтический эффект вне зависимости от статуса TP53, в том числе через активацию p38MAPK [58]. Комбинирование ингибиторов PPM1D с антагонистами MDM2, предотвращающими деградацию p53, продемонстрировало синергетический эффект на доклинических моделях ОМЛ с TP53 дикого типа, дополнительно усиливая апоптотическую сигнализацию и подавляя рост лейкозных клеток [57]. Представление о том, что PPM1D является весьма перспективной терапевтической мишенью при ОМЛ, убедительно подтверждается обширными доклиническими данными, так как повышенная экспрессия PPM1D коррелирует с агрессивным течением заболевания, химиорезистентностью и неблагоприятными клиническими исходами как при первичном ОМЛ, так и при ОМЛ, связанном с терапией (рис. 2) [11, 13, 14, 22, 28]. Хотя мутации PPM1D встречаются реже, чем другие мутации, приводящие к развитию ОМЛ, их влияние на резистентность к терапии непропорционально велико. Способность PPM1D подавлять реакцию клеток на DDR и способствовать выживанию в условиях химиотерапевтического стресса делает его критически важным звеном в сигнальной сети, определяющей судьбу лейкозных клеток [14, 57]. В отличие от традиционных химиотерапевтических препаратов, механизмы действия которых в целом цитотоксические и неселективные, таргетные методы, такие как ингибирование PPM1D, обладают потенциалом для более селективной эрадикации лейкозных клеток, не затрагивая при этом нормальные гемопоэтические клетки-предшественники.
Возможные недостатки PPM1Dв качестве мишени для терапии
Как упоминалось ранее, фосфатаза PPM1D экспрессируется в нормальных тканях организма
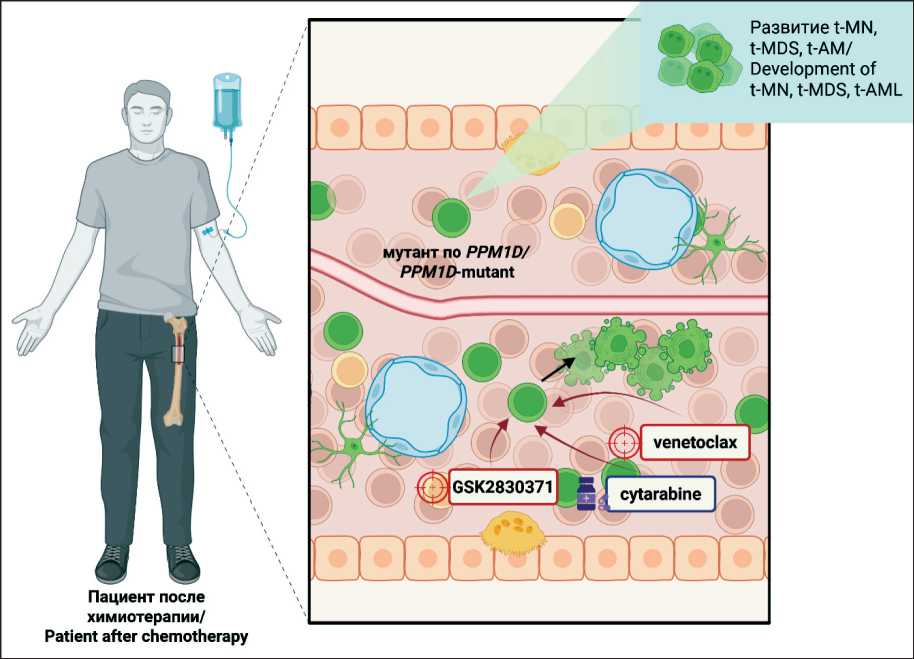
Рис. 2. Появление клонов ГСК с мутациями в гене PPM1D после химиотерапии может привести к развитию t-MN (t-MDS и t-AML). Комбинирование химического ингибитора PPM1D, таргетной терапии и химиотерапии, направленной на клетки с мутацией в гене PPM1D , может способствовать преодолению резистентности к терапии и улучшению исхода терапии.
Примечание: created in BioRender. Pukhalskaya, T. (2025)
Fig. 2. The emergence of hematopoietic stem cell clones harboring mutations in the PPM1D gene following chemotherapy may contribute to the development of t-MN (t-MDS and t-AML). The combination of a small-molecule PPM1D inhibitor with targeted therapy and chemotherapy directed against PPM1D-mutant cells may facilitate the overcoming of treatment resistance and improve overall therapeutic outcomes. Note: created in BioRender. Pukhalskaya, T. (2025)
в ответ на активацию p53, где она регулирует клеточный ответ на стресс [7, 14, 25–28]. Традиционно предполагалось, что подавление активности PPM1D приводит к активации p53-зависимых сигнальных путей, поскольку эта фосфатаза является негативным регулятором p53, поэтому в опухолях с мутантным или делецированным TP53 такая стратегия ингибирования считалась неэффективной. Например, показано, что в нейробластоме с интактным p53 GSK2830371 подавляет рост клеток с дикой формой p53, но не действует на клетки с мутантным p53 [25]. Однако последние работы существенно пересмотрели эту парадигму. Это связано с тем, что фосфатаза PPM1D регулирует целый ряд p53-независимых сигнальных путей, поэтому ее ингибирование останавливает рост даже тех опухолей, в которых TP53 мутирован или полностью инактивирован [59]. В клетках с делецией TP53 фосфатаза PPM1D лишена своего основного субстрата, что может способствовать расширению спектра ее мишеней за счет взаимодействий с альтернативными субстратами. Избыточная экспрессия фосфатазы PPM1D в опухолевых клетках с инактивированным p53 опосредует RUNX2-зависимую активацию апоптоза в ответ на противоопухолевую терапию через дефосфорилирование Ser432 RUNX2 и по- следующую трансактивацию промотора гена Bax, тогда как в нормальных тканях с диким типом Tp53 PPM1D подавляет гиперактивацию p53 и защищает от цитотоксических побочных эффектов химиотерапии [60].
В контексте влияния подавления PPM1D в нормальных клетках организма было показано, что химическое ингибирование PPM1D не приводит к значительным токсическим эффектам [61]. Генетический нокаут Ppm1d у мышей приводил к дефектам T- и B-клеточного ответа и дефектам репродуктивной системы [25, 61]. При этом делеция Ppm1d не сказывается на продолжительности жизни мышей. Также показана роль PPM1D в развитии нейтрофилов [62]. Мыши с делецией гена Ppm1d имели повышенное количество нейтрофилов в крови и КМ с признаками ускоренного созревания [62]. В целом, генетическая делеция Ppm1d не несет тяжелых последствий, но данные свидетельствуют о том, что PPM1D играет роль в дифференцировке клеток иммунной системы [62].
Сейчас GSK2830371 предлагается в качестве основного химического соединения для ингибирования PPM1D, однако существуют и другие молекулы [35, 61]. GSK2830371 связывается с аллостерическим сайтом PPM1D с высокой аффинностью, что делает его высокоселективным ингибитором [21, 61]. Важно отметить, что исследования показали, что GSK2830371 обладает пероральной биодоступностью in vivo [61]. Однако относительно короткий период полувыведения GSK2830371 может ограничивать его клиническое применение [61]. Дальнейшая модификация GSK2830371 в качестве ведущего соединения, как ожидается, позволит разработать низкомолекулярный ингибитор PPM1D с улучшенными фармакокинетическими свойствами.
Несмотря на убедительные доклинические данные, ингибиторы PPM1D пока не дошли до поздних стадий клинических испытаний. Наиболее изученные соединения, такие как GSK2830371, остаются на доклиническом этапе. Отсутствие зарегистрированных препаратов и валидированных биомаркеров ответа ограничивает практическое внедрение данной стратегии.
Заключение
Острый миелоидный лейкоз характеризуется высокой агрессивностью течения и неблагоприятным прогнозом, особенно у пожилых пациентов, у которых 5-летняя общая выживаемость не превышает 10–20 % [63]. Нарастающее количество
