Роль MTOR в репрограммировании опухоль-ассоциированных макрофагов и в канцерогенезе (обзор)
")
Автор: Мартынова Т.Н., Малышев И.Ю.
Журнал: Саратовский научно-медицинский журнал @ssmj
Рубрика: Патологическая физиология
Статья в выпуске: 1 т.16, 2020 года.
Бесплатный доступ
Макрофаги - гетерогенная популяция клеток, которые дифференцируются в различные функциональные группы в зависимости от полученных сигналов. Агонисты toll-like рецепторов и цитокины воздействуют на соответствующие рецепторы, вызывая изменения метаболизма и экспрессии генов в миелоидных клетках системы врожденного иммунитета. Данные изменения в фагоцитах происходят в процессе их активации, в результате они становятся поляризованными в том или ином направлении, приобретая определенный фенотип. Данные изменения экспрессии генов реализуются в опухолевом микроокружении. mTOR (mammalian target of rapamycin) - серин/треониновая протеинкиназа, которая реагирует на различные стимулы окружения клетки изменением ее метаболизма. mTOR также принимает участие в процессе активации макрофагов. Тема представленного обзора литературы: роль mTOR в репрограммировании макрофагов, в том числе опухоль-ассоциированных. Для поиска информации использовалась база данных PubMed.
Макрофаги, опухоль, репрограммирование
Короткий адрес: https://sciup.org/149135498
IDR: 149135498 | УДК: 616.092
The role of MTOR in reprogramming of tumor associated macrophages and in cancerogenesis (review)
Macrophages are heterogeneous population of cells, which differentiate into distinct functional groups depending of the external signals. Agonists of toll-like receptors (TLRs) and cytokines bind to corresponding receptors and cause changes in the metabolism and expression of genes in myeloid cells located in the innate immune system. These changes in phagocytes are occurring during their activation. As a result, they become polarized, acquiring a determined phenotype. This gene expression changes realize in the tumor microenvironment. mTOR - serine/threonine protein kinase, which receives various signals from cell environment. It causes changes in protein biosynthesis and cell metabolism. mTOR takes part in the activation of macrophages influenced by cytokines or agonists of TLRs. It determines the role of kinase in reprogramming mechanisms and in the regulation of cytokines production according to the type of received signals respectively. The review has been made about the role of mTOR in macrophages' reprogramming, in particular tumor-associated ones. The PubMed resource is used for information search
Текст научной статьи Роль MTOR в репрограммировании опухоль-ассоциированных макрофагов и в канцерогенезе (обзор)
а также принимают участие в поддержании механизмов гомеостаза, таких как тканевое ремоделирование и заживление раны [1].
Клетки системы врожденного иммунитета обеспечивают иммунологическую память через эпигенетическое репрограммирование после стимуляции с микробными лигандами. Данная функция позволяет усиливать неспецифический воспалительный ответ после вторичной стимуляции — процесс, называемый «обучением иммунитета». Были идентифицированы эпигеномные копии «обученных» моноцитов, что выявило несколько важных иммунологических и метаболических механизмов, которые лежат в основе этих изменений. Интересно, что подобное долговременное репрограммирование, приводящее к продукции цитокинов, индуцировано эндогенными DAMPs (молекулярными образами, ассоциированными с повреждениями). Обнаружено, что эндогенные сигналы, ассоциированные с тканевым повреждением и асептическим воспалением, могут индуцировать «тренированный» («обученный») иммунитет через эпигенетическую регуляцию транскрипционных программ и что постоянно воздействующие DAMPs влияют на ход воспалительного процесса, а сигналы тканевого происхождения являются критичными для регуляции выраженности и типа иммунного ответа, производимого организмом [2]. В обученных макрофагах промоторы генов, кодирующих компоненты путей иммунного сигналинга, активированы.
То же самое происходит в опухоли: сигналы, полученные в ее микроокружении, начинают программировать макрофаги, мигрировавшие в опухоль для борьбы с ней. Так появляется множество популяций макрофагов: супрессорные клетки миелоидного происхождения, регуляторные Т-клетки и опухоль-ассоциированные макрофаги (ОАМ), которые существуют в опухолевом микроокружении и поддерживают рост опухоли и напрямую, и опосредованно [3].
mTOR (первично «TOR млекопитающих», в настоящее время –«механистический TOR») — ключевой регулятор клеточного метаболизма, который функционирует как питательный/энергетический сенсор и регулирует метаболические процессы синтеза белка, гликолиза и липогенеза в зависимости от доступности питательных веществ [4]. Выявлено нарушение регуляции mTOR-сигналинга в таких заболеваниях, как рак, ожирение, диабет 2-го типа, нейродегенеративные заболеваниях, что говорит об актуальности исследования данного вопроса.
mTOR принимает участие в процессах актива-ции/поляризации макрофагов в зависимости от получаемых стимулов [5], тем самым играя заметную роль в модуляции иммунного ответа.
Роль макрофагов в иммунном ответе: концепция поляризации. Опухоли развиваются как экосистемы, состоящие из опухолевых, стромальных и инфильтрирующих строму иммунных клеток. Макрофаги являются главными компонентами этой системы [6]. Однако популяция этой системы неоднородна.
Моноциты, макрофаги и дендритные клетки в совокупности образуют систему мононуклеарных фагоцитирующих клеток — гетерогенную популяцию клеток, которые могут дифференцироваться в отдельные функциональные подгруппы, формирующиеся при помощи сигналов, полученных от микроокружения [7]. Термин «активация» означает преобразование макрофагов под воздействием экзогенных агентов (цитокинов или агонистов рецепторов (TLR)), что приводит к определенным образам экспрессии генов; многие используют в том же контексте обозначение «поляризация».
Чтобы выполнять различные функции, макрофаги приобретают специфические фенотипы, которые могут быть охарактеризованы определенным видом экспрессии генов, образом поверхностных молекул и продукцией биологических медиаторов и метаболитов [8]. На границах данного континуума поляризационного статуса макрофагов находятся два фенотипа, которые могут быть определены как М1-провоспалительный/противоопухолевый против М2-противовоспалительного/проопухолевого.
Данные разновидности макрофагов принимают соответствующий образ экспрессии генов в зависимости от стимулов, полученных в условиях микроокружения.
Например, в 2011 г. Krausgruber и коллеги показали, что IRF5 является критическим белком для M1 макрофагальной поляризации. И GM–CSF, и IFN-γ стимулы индуцируют IRF5-экспрессию, которая напрямую активирует 20 M1-специфичных генов и ингибирует 19 M2-специфичных генов, кодирующих цитокины [9]. IRF5 вовлечен в М1-поляризацию, индуцируя транскрипцию p40 субъединицы интерлейкина 12 (IL-12p40), IL-12p35 и IL-23p19, а также снижая транскрипцию IL-10 [9].
Таким образом, M1-макрофаги под специфическими условиями усиливают воспалительный процесс, что может стать угрожающим для состояния здоровья. Но в опухоли, особенно на поздних стадиях развития, складывается обратная ситуация, когда к опухолевым клеткам формируется толерантность. В данном случае немаловажную роль играют ОАМ.
Макрофаги, активированные через путь, противоположный классическому, относятся к M2, или альтернативному пути. Было выявлено, что такие стимулы, как CSF-1, IL-4, IL-10, TGF-β и IL-13, грибковая и гельминтная инфекция управляют M2-субпопуляцией поляризации, индуцируя IL-10 в высокой концентрации и IL-12 в низкой [10].
Транскрипционные факторы PPARγ и PPARδ активируются при помощи STAT6 и необходимы для M2-поляризации [11].
Экспериментально доказано, что среди ОAM M2-подобные макрофаги поддерживают в опухоли инициацию, прогрессию и выживаемость; они ингибируют иммуностимулирующие сигналы и не имеют цитотоксической активности. ОAM-инфильтрация в опухоли коррелирует с неблагоприятным прогнозом [12]. Более того, многочисленные исследования показали, что ОАМ главным образом ответственны за резистентность к классической противоопухолевой терапии (лучевой и химиотерапии), а также ограничивают эффективность методов иммунотерапии (например, анти-PD1) [13].
ОАМ собираются в гипоксических областях; они являются MHClow, имеют проангиогенные свойства и плохую антигенпрезентирующую способность; с другой стороны, макрофаги, локализованные в областях нормоксии, могут быть более гетерогенными, и некоторые из них имеют М1-фенотип с MHChigh-экспрессией [14].
Многочисленные свидетельства подтверждают, что в опухолях мыши и человека ОАМ главным образом проявляют альтернативно активированный фенотип, который ассоциирован с поддержкой роста опухоли, ремоделированием внеклеточного матрикса, ангиогенезом и супрессией адаптивного иммунитета [15].
M2-представительство в опухолевом микроокружении также поддерживает выживаемость, рост опухоли и метастазирование [16].
Опухолевые и иммунные клетки в соответствующем микроокружении продуцируют цитокины, факторы роста и метаболиты, которые поддерживают про-опухолевую поляризацию ОАМ. Медиаторы, такие как CSF-1, CCL2 и VEGF, поддерживают вовлечение ОАМ в микроокружение опухоли [8]. Th2-цитокины IL-4, IL-13, IL-10 и TGFβ, продуцируемые Treg и ТАМ, являются ключевыми направляющими иммуносупрессии.
Далее будет рассмотрена роль mTOR в модуляции сигналов в ответ на Th1- и Th2-цитокины, а следовательно, и в репрограммировании макрофагов. Фосфатидилинозитол-3-киназный (PI3K) сигнальный путь, который активирует множественные сигнальные каскады через продукцию вторичного посредника PIP3, регулирует выживаемость макрофагов и экспрессию генов через активацию семейства Akt серин/треонинпротеинкиназ. PI3K/Akt-сигнальный путь контролирует активацию mTOR [17].
Что такое mTOR? mTOR (mammalian target of rapamycin) — атипичная серин/треонинпротеинки-наза, принадлежащая к PIKK (PI3K) семейству киназ с молекулярным весом около 290 kDa.
mTOR — ключевой каталитический компонент, локализующийся в отдельных субклеточных компар-тментах (лизосомах, митохондриях, цитоплазматической мембране, эндоплазматическом ретикулуме, ядре) и состоящий из двух структурно и функционально отдельных комплексов mTORC1 и mTORC2 [4] (рис. 1).
mTORC1-путь объединяет входы, как минимум, от пяти главных внутриклеточных и внеклеточных стимулов: факторов роста, стресса, энергетического состояния, кислорода и аминокислот, опосредуя многие базовые процессы, включая синтез белка и липидов и аутофагию. mTORC1 отвечает на различные стимулы микроокружения, контролируя многие процессы по производству/использованию энергии и питательных веществ. mTOR-сигналинг влияет на главнейшие функции клетки, регулируя основные виды поведения клетки: рост (увеличение массы) и пролиферацию [4].
Немало известно о том, как данные стимулы регулируют mTORC1-сигналинг, хотя присутствуют сведения о связи тех же стимулов с mTORC2 [18].
PI3K-mTOR-путь. Различные рецепторные тиро-зинкиназы активируют PI3K через отдельные стыковочные белки, такие как FRS (FGF Receptor Substrate) или GAB (c-Met or EGFR), или через прямое связывание PI3K (Platelet-derived Growth Factor Receptor).
Инсулин и IGF1 присоединяется к своим распознающим рецепторным тирозинкиназам (RTKs), после активации которых происходит вовлечение в процесс PI3-киназы, а затем ее регуляторной (p85) и каталитической (p110) субъединиц, что приводит к фосфорилированию фосфатидил-4,5-бифосфата (PIP2) и превращению его в фосфатидил-3,4,5-трифосфат (PIP3). PTEN (Phosphataseandtensinhomologuedelete donchromosome 10) катализирует обратную реакцию, обеспечивая инактивацию Akt в покоящихся клетках. PIP3 привлекает киназу PDK1 (phosphoinositide-dependentkinase 1) к плазматической мембране и активирует ее фосфорилированием. Активированная PDK1 затем фосфорилирует Akt и PKCθ. Akt фосфорилирует TSC2 в многочисленных сайтах, напрямую связывая PI3K-Akt с TSC-mTORC1 [18].
TSC2 млекопитающих увеличивает внутреннюю скорость GTP гидролиза Rheb, превращая Rheb из GTP-связанной (активной) в GDP-связанную (неактивную) форму, которая напрямую взаимодействует с mTORC1 и мощно стимулирует его киназную активность.
ФосфорилированиеTSC2 при помощи Akt снижает TSC2 GAP активность в отношении Rheb, что ин-
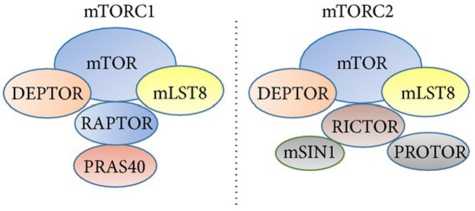
Рис. 1. mTORC1 и mTORC2:
mTORC1 включает в себя собственно mTOR (каталитическая субъединица), RAPTOR, DEPTOR, PRAS40, mLST8; mTORC2 состоит из mTOR, RICTOR, DEPTOR, PROTOR1 /2, mSIN1 и mLST8
дуцирует прямую активацию mTORC1 [18]. mTORC1 фосфорилирует 4E-BP (eukaryoticinitiationfactor 4E (eIF4E) — bindingprotein) и S6K (p70 ribosomalS6 Kinase), что приводит к изменению биогенеза рибосом и трансляции белков [18] (рис. 2).
Инсулин и IGF1 присоединяется к рецепторным тирозинкиназам (RTKs), что приводит к аутофосфорилированию рецептора. После активации рецепторной тирозинкиназы происходит вовлечение в процесс PI3-киназы, а затем ее регуляторной (p85) и каталитической (p110) субъединиц, что приводит к фосфорилирилированию фосфатидил-4,5-бифосфата (PIP2) и превращению его
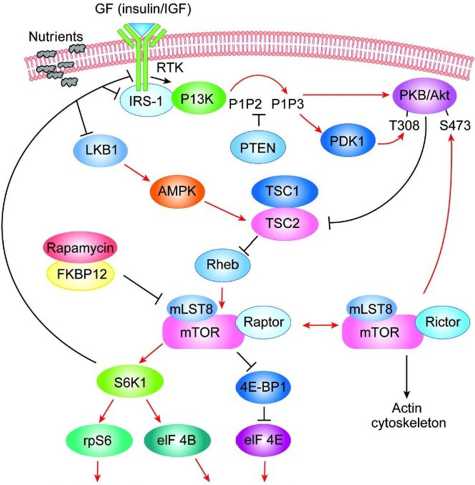
Биогенез рибосом Кэп-зависимая трансляция (циклин D1, с-Myc, HIF-1α, EGF)
Рис. 2. PI3K-mTOR-сигналинг:
GF (факторы роста) — инсулин, инсулиноподобный фактор роста; RTK — рецепторная тирозинкиназа; IRS-1 — инсу-линрецепторный субстрат-1; PI3K — фосфатидилинозитол-3киназа; PKB (AKT) — протеинкиназа В; PDK1 — фосфои-нозитид-зависимая киназа; LKB1 — печеночная киназа 1; PTEN — фосфатаза и тензин-гомолог, удаленный на 10-й хромосоме; AMPK — AMФ-активируемая протеинкиназа;
TSC1 /2 — комплекс Tuberoussclerosis; Rheb — Ras-гомолог, питающий головной мозг; FKBP12 — FK506-связывающий белок; mTORC1: mTOR, Raptor, DEPTOR, PRAS40, mLST8; mTORC2: mTOR, Rictor, DEPTOR, protor1/2, mSinl, mLST8;
S6K1 — S6-киназа 1; rpS6 — рибосомальный белок S6; eIF4B — эукариотический фактор инициации трансляции 4В; 4E-BP1 — eIF4E-связывающий белок; eIF4E — эукариотический фактор инициации трансляции 4Е в фосфатидил-3,4,5-трифосфат (PIP3). PIP3 привлекает киназу PDK1 (phosphoinositide-dependent kinase 1) к плазматической мембране и фосфорилирует ее, которая, в свою очередь, активирует Akt и PKCθ. Akt фосфорилирует TSC2. ИнактивированныйTSC2 аналогичным образом действует на Rheb, в результате чего активируется mTORC1, 4E-BP (eukaryotic initiation factor 4E (eIF4E) — binding protein) и S6K (p70 ribosomal S6 Kinase).
mTOR-активация в макрофагах:
-
1. Роль mTOR в TLR4-сигналинге. Как известно, TLR4-сигналинг—один из наиболее часто задействованных путей в воспалительном процессе, а хроническое воспаление в ряде случаев выступает пусковым моментом в канцерогенезе. По данным Schmitz, ингибирование mTOR снижает TLR-индуцированное высвобождение IL-10, TNF-a, IL-6 и оксида азота макрофагами и дендритными клетками, а также усиливает процессинг каспазы-1 и таким образом повышает продукцию биоактивного IL-1β [19].
-
2. Активация Nod2-рецепторов. Nod2-стимуляция при помощи мурамилдипептида (MDP) приводит к фосфорилированию p70 S6 (субстрата mTOR) [20] и Akt, вышестоящего активатора mTOR. Активация mTOR необходима для последующей секреции IL-10, TGF-β и IL-1Ra, которая лежит в основе Nod2-опосредованной толерантности [21].
-
3. IFN-γ-сигналинг. Под воздействием IFN-γ mTOR снижает экспрессию с/ЕВРв и MARCO, ингибируя таким образом неопсонизированный фагоцитоз. IFN-γ индуцировал повышенную активность mTOR и снижал экспрессию с/ЕВРв (CCAAT enhancer-binding protein β) в макрофагах. А рапамицин значительно отменял ингибиторный эффект IFN-y на неопсонизи-рованный фагоцитоз в макрофагах и восстанавливал экспрессию с/ЕВРв и MARCO [22].
-
4. IL-27. Экзогенный IL-27 индуцирует активацию mTOR через JAK/PI3K-путь и ингибирует IFN-y- стимулированную аутофагию. IL-27 также повышает экспрессию Mcl-1 через PI3k-путь [23] посредством конкурентной активации JAK/PI3 K/Akt/mTOR-каскада, так же как и увеличения Mcl-1, ингибирует IFN-γ-индуцированную аутофагию и элиминацию внутриклеточных бактерий в макрофагах [6].
-
5. Под воздействием минимально модифицированных LDL (mmLDL) активность mTORC1 усиливает индукцию хемокинов, усиливая IL6-сигналинг [25].
В то же время активация Pl3к/Akt-пути снижает продукцию IL27, так как фармакологическая блокада фосфорилирования Akt приводит к усиленной продукции IL-27 (p28) в LPS-стимулированных макрофагах [24].
В классическом IL6-сигналинге соединение цитокина с его рецептором IL6Ra на клеточной поверхности приводит к вовлечению сигнального передатчика gp130 и активации JAK1, которая фосфорилирует STAT3. Фосфорилированный STAT3 димеризуется и идет в ядро, где он начинает свою транскрипционную программу [26].
Несколько исследований подтвердили, что mTORC1 ответствен за фосфорилирование STAT3 в Ser [27]. Ding A. и коллеги обнаружили, что фосфорилирование STAT3 Ser727 в ответ на mmLDL было снижено в Mac-RapKO-макрофагах, и предварительное воздействие рапамицина отменяло фосфорилирование S6 и STAT3 Ser727. Фосфорилирование в Ser727 STAT3 усиливало его способность противодействовать эффекту BCL-6 в Ccl2-промоторе [25].
Стимуляция макрофагов с mmLDL индуцировала экспрессию хемокиновых генов Ccl2, Ccl3 и Ccl7 [28].
mTORCI усиливал эффект IL6/STAT3 на Ccl2-экспрессию генов, что обнаруживает новый перекрест сигналов между воспалительными путями, индуцированными mmLDL, и активацией mTORC1, индуцирующей хемотаксис моноцитов в очаг опухоли.
Много работ выполнено по определению роли растворимых факторов, таких как хемокины и факторы роста, в поляризационной функции макрофагов на протяжении опухолевой прогрессии. Например, хемокин CCL2 и макрофагальный колониестимулирующий фактор вовлекают моноциты в ткань опухоли, а затем IL-10 (при участии mTOR), IL-4, IL-13 и другие цитокины в микроокружении стимулируют моноциты к дифференцировке в M2 ОАМ [29].
mTOR в ОАМ. Для начала проясним роль mTOR в репрограммировании макрофагов в сторону М2-поляризации.
Под действием LPS в макрофагах mTORC1 активирует транскрипцию и трансляцию HIF1α [30], который, в свою очередь: а) связывается с TLR4-промотором, так же как с VEGF-промотором [31]; б) индуцирует гипоксия-ответственные гены, вовлеченные в ангиогенез и метаболизм глюкозы (например, фактор роста эндотелия (VEGF), глюкозные транспортеры (GLUT1 и GLUT3) и гексокиназы (HK1 и HK2), PAI-1, а также iNOS и многие другие, кодирующие ферменты гликолиза [32]; MMP-2 (матриксную металлопротеиназу-2) [33].
Более того, mTORCI связан с глутаминовым метаболизмом через контроль гидролиза глутамина до глутамата (критический момент в утилизации глутамина) [5]. Даже М1-макрофаги совмещают потребление глутамина с анаплеротическим пополнением цикла Кребса, который поддерживает продукцию цитрата и стабилизацию HIF1α [5].
Таким образом, mTOR обеспечивает не только трансляцию HlF-1a, но и ряда ферментов, поддерживающих активность гликолиза в опухоли и феномен Warburg, а усиленный гликолиз управляет потоком через ПФП, что приводит к повышению образования НАДФ, необходимого для синтеза АФК.
c-Myc усиливал IL-4-опосредованную STAT6-активацию и повышенную экспрессию 45% генов, коррелирующих с альтернативной активацией макрофагов. mTOR (S6K) поддерживает активацию c-Myc путем фосфорилирования в Ser145 Mad1, ингибитора c-Myc [35].
-
IL -4 и IL-13 сигнализируют через IRF/STAT, активируя STAT6 в М2-макрофагах. STAT6 индуцирует экспрессию регуляторов транскрипции, таких как PPAR-Y [36]. mTORCI поддерживает экспрессию и активность фактора PPAR-γ (peroxisome proliferator-activated receptor γ), который необходим для поддержания метаболического сдвига в сторону окислительного фосфорилирования, и поддерживает М2-экспрессию генов (Arg1), усиливая эффекторный фенотип M2-макрофагов (синтез коллагена) [37], а также Chi3l3, Mrc1 и Jag1).
-
IL-4 индуцирует также экспрессию PGC-1β мРНК и белка в покоящихся и в активированных макрофагах [11].
Из вышеизложенного следует, что mTOR необходим для осуществления метаболического сдвига и формирования М2-фенотипа макрофагов.
Стимуляция при помощи факторов роста (инсулина, GM–CSF и IL-3) приводит к STAT3-фосфорилированию в Ser727. Активированный STAT3 незаменим для поддержания клеточной выживаемости через регуляцию экспрессии Mcl-1 [38].
Известно, что многие целевые гены STAT3 вовлечены в развитие раковой опухоли, например HIF-1α, который играет ведущую роль в ангиогенезе [39], и TGFβ1, который поддерживает рост опухолевых клеток и метастазирование [40].
По данным Wei Chen и коллег, mTOR регулирует способность макрофагов индуцировать ангиогенез под действием IL10 : в клетках иммунной системы и в опухолевых клетках mTOR увеличивает секрецию IL-10, который поддерживает продукцию VEGF (а ра-памицин оказывает обратный эффект на секрецию IL-10 и VEGF) [41] .
Активированный mTOR поддерживает ангиогенез, индуцированный макрофагами, также через STAT3 (т. к. STAT3 является нижестоящей целью mTOR в макрофагах и других клеточных типах) [42].
Сведения, полученные Dru S. Dace и коллегами, также подтверждают роль IL10/STAT3-сигналинга в экспрессии VEGF: макрофаги, стимулированные с IL-10 в течение 10 минут или гипоксией в течение 24 часов, демонстрировали повышенное фосфорилирование STAT3 по сравнению с исходным нормоксиче-ским уровнем. Было показано, что STAT3-активация и сигналинг может приводить к экспрессии VEGF [43].
STAT3 может быть активирован через JAK/STAT3 (pY705) — путь, который индуцирует экспрессию HIF-1а и vEgF MEK/ERKMAPK-путь управляет активацией STAT3 (pS727), и c-KIT/PI3K/AKT/mTOR-путь также управляет фосфорилированием STAT3 (pS727), которое приводит к экспрессии IDO и HIF-1α [44].
Как упоминалось выше, активация mTOR под воздействием LPS увеличивает экспрессию IL10 [19].
IL10, в свою очередь, активирует STAT3 [45].
Активация сигнального передатчика и активатора транскрипции 3 (STAT3) опосредует роль ОAM в ангиогенезе при помощи повышения экспрессии нескольких проангиогенных факторов, например bVEGF и bFGF [46].
Доказано, что MMP-2 (один из целевых генов HIF1α, а следовательно, результат активации mTOR) и MMP-9 стимулируют продукцию TGFβ, VEGF, PDGF и FGF, а их экспрессия коррелирует с повышенной инвазивностью опухоли и плохим прогнозом [12]. Таким образом, проявляется роль mTOR в индукции перечисленных выше факторов роста в опухолевой прогрессии.
Сходный механизм описан для урокиназа-подоб-ного активатора плазминогена (uPA), который после соединения со своим рецептором (uPAR) на ОAM индуцирует расщепление плазминогена в плазмин, что приводит к деградации внеклеточного матрикса с последующим выделением факторов роста и неблагоприятным исходом [47].
Под воздействием PSGL-1 активация mTOR-сигналинга приводит к повышению трансляции рецептора uPAR (урокиназного активатора плазминогена), поддерживающего адгезию, миграцию и хемотаксис макрофагов в очаг повреждения.
Ключевым игроком в метастазировании является TGFβ, который продуцируется ОАМ; TGFβ запускает эпителиально-мезенхимальный переход, сдвигающий эпителиальные раковые клетки в сторону мезенхимального фенотипа, более склонного к миграции [48]. Иммуносупрессивная функция ОАМ также опосредуется при помощи секреции TGFβ вместе с IL-10, который снижает CD8+ T-клеточные функции не только прямой транскрипционной репрессией генов, кодирующих такие субстанции, как перфорины, гранзимы и цитотоксины, но и опосредованно, путем стимуляции пролиферации Treg-клеток или путем супрессии противоопухолевых функций дендритных клеток [49]. Более того, ОАМ индуцируют старвацию аминокислот в Т-клетках через продукцию аргиназы и индоламин-2,3-диоксигеназы (IDO) [50].
Заключение. Макрофаги представляют собой гетерогенную популяцию клеток, которые обладают удивительной пластичностью и живо реагируют на изменения в системе микроокружения. IFN-γ, LPS и GM–CSF индуцируют приобретение макрофагом свойств и метаболизма, характерных для М1-фенотипа (в том числе секреции провоспа-лительных цитокинов), а CSF-1, IL-4, IL-10, TGF-β и IL-13 индуцируют приобретение макрофагом М2-характеристик. В опухоли присутствуют и М1-, и М2-подобные макрофаги. Однако по мере опухолевой прогрессии ОАМ начинают приобретать все больше М2-характеристик, что в дальнейшем обусловливает неблагоприятный прогноз.
mTOR активно включается в процесс репрограммирования макрофагов в зависимости от вида стимула: под воздействием LPS киназа обеспечивает переход к гликолитическому окислению, которое лежит в основе метаболического сдвига, в том числе за счет повышения экспрессии и активации Hif-1α. Hif-1α, в свою очередь соединяясь с целевыми генами, принимает участие в поддержании процесса хронического воспаления и в канцерогенезе. Под воздействием IL4,13 mTOR-сигналинг обеспечивает экспрессию и активность PPAR-γ и PGC-1β, факторов транскрипции, необходимых для поддержания окислительного метаболизма в М2-макрофагах, экспрессии их маркеров, а также секрецию противовоспалительного цитокина IL-10, который также незаменим в процессе канцерогенеза.
mTORC1 вступает во взаимодействие с другими сигнальными путями в макрофагах:
-
- в TLR4-сигналинге активация mTOR способствует продукции IL-10, TNF-α, IL-6;
-
- в Nod2-сигналинге — продукции IL-10, TGF-β и IL-1Ra;
-
- под воздействием IFN-γ mTOR снижает экспрессию с/ЕВРв и MARCO, ингибируя неопсонизиро-ванный фагоцитоз;
-
- IL-27 индуцирует активацию mTOR и ингибирует IFN-γ-стимулированную аутофагию;
-
- под воздействием минимально модифицированных LDL (mmLDL) активность mTORC1 усиливает индукцию хемокинов CCL2, -3, -7, усиливая IL6-сигналинг, что обусловливает хемотаксис моноцитов в очаг опухоли, которые затем пополняют ряды ОАМ;
-
- факторы роста (инсулина, GM–CSF и IL-3) приводят к STAT3-фосфорилированию, что повышает выживаемость клеток, в том числе опухолевых, повышает продукцию bVEGF и bFGF, а также TGFβ1, принимающего участие в процессах роста и метастазирования.
mTOR в макрофагах принимает участие в продукции IL-10 под воздействием различных стимулов (сигналы с TLR4, Nod2-рецепторов). IL-10, в свою очередь, активирует STAT3-сигналинг, что приводит к экспрессии факторов HIF-1α, VEGF, FGF, а также стимулированию выработки TGFβ. Продукция данных цитокинов при непосредственном (IL-10) и косвенном участии mTOR (TGFβ, MMP2, VEGF, PDGF,
FGF) обусловливает роль киназы в повышении выживаемости опухолевых клеток, росте опухоли, в том числе инвазивном, ангиогенезе, метастазировании и неблагоприятном прогнозе заболевания при ее активации в ОАМ, что следует принимать во внимание при терапевтическом подходе в вопросе опухолей.
Список литературы Роль MTOR в репрограммировании опухоль-ассоциированных макрофагов и в канцерогенезе (обзор)
- Katholnig K, Linke M, Pham H, et al. Immune responses of macrophages and dendritic cells regulated by mTOR signaling. Biochemical Society Transactions 2013; 41 (4): 927-33.
- Crisan TO, Netea MG, Joosten LA. Innate immune memory: Implications for host responses to damage-associated molecular patterns. European Journal of Immunology 2016; 46 (4): 817-28.
- Luca C, Fragkogianni S, Sims AH, et al. Human Tumor-Associated Macrophage and Monocyte Transcriptional Landscapes Reveal Cancer-Specific Reprogramming, Biomarkers, and Therapeutic Targets. Cancer Cell 2019; 35 (4): 588-602.
- Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell 2012; 149 (2): 274-93.
- Covarrubias AJ, Aksoylar HI, Horng T. Control of macrophage metabolism and activation by mTOR and Akt signaling. Seminars in Immunology 2015; 27 (4): 286-96.
- Abdalla AE, Li Q, Xie L, et al. Biology of IL-27 and its Role in the Host Immunity against Mycobacterium Tuberculosis. International Journal Biological Sciences 2015; 11 (2): 168-75.
- Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo Veritas. The Journal of Clinical Investigation 2012; 122 (3) 787-95.
- Biswas SK. Metabolic Reprogramming of Immune Cells in Cancer Progression. Immunity 2015;43: 435-49.
- Krausgruber T, Blazek K, Smallie T, et al. IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nature Immunology 2011; 12 (3): 231-8.
- Jenkins SJ, Ruckerl D, Thomas GD, et al. IL-4 directly signals tissue-resident macrophages to proliferate beyond homeostatic levels controlled by CSF-1. The Journal of Experimental Medicine 2013; 210 (11): 2477-91.
- Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature 2007; 447 (7148): 1116-20.
- Mantovani A, Marchesi F, Malesci A, et al. Tumor-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol 2017; 14: 399-416.
- Allavena P, Mantovani A. Immunology in the clinic review series; focus on cancer: Tumor-associated macrophages: Undisputed stars of the inflammatory tumor microenvironment. Clin Exp Immunol 2012; 167: 195-205.
- Zumsteg A, Christofori G. Corrupt policemen: Inflammatory cells promote tumor angiogenesis. Curr Opin Oncol 2009; 21: 60-70.
- Lawrence T, Natoli G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol 2011 Oct 25; 11 (11): 750-61.
- R. Ostuni F, Kratochvill PJ, Murray, et al. Macrophages and cancer: from mechanisms to therapeutic implications. Trends in Immunology 2015; 36 (4): 229-39.
- Mercalli A, Calavita I, Dugnani E, et al. Rapamycin unbalances the polarization of human macrophages to M1. Immunology 2013; 140 (2): 179-90.
- Sengupta S, Peterson TR, Sabatini DM. Regulation of the mTOR complex 1 pathway by nutrients, growth factors, and stress. Molecular Cell 2010; 40 (2): 310-22.
- Schmitz F, Heit A, Dreher S, et al. Mammalian target of rapamycin (mTOR) orchestrates the defense program of innate immune cells. The European Journal of Immunology 2008; 38 (11): 2981-92.
- Chen H, Cowan MJ, Hasday JD, et al. Tobacco smoking inhibits expression of proinflammatory cytokines and activation of IL-1R-associated kinase, p38, and NF-kappaB in alveolar macrophages stimulated with TLR2 and TLR4 agonists. Journal of Immunology 2007; (179): 6097-106.
- Hedl M, Abraham C. Secretory Mediators Regulate Nod2-Induced Tolerance in Human Macrophages. Gastroenterology 2011; 140 (1): 231-41.
- Wang Z, Zhou S, Sun C, et al. Interferon^ inhibits nonopsonized phagocytosis of macrophages via an mTORC1-c/EBPß pathway. Journal of Innate Immunity 2015; 7 (2): 165-76.
- Sharma G, Dutta RK, Khan MA, et al. IL-27 inhibits IFN-y induced autophagy by concomitant induction of JAK/PI3 K/Akt/mTOR cascade and up-regulation of Mcl-1 in Mycobacterium tuberculosis H37Rv infected macrophages. International Journal of Biochemistry & Cell Biology 2014; (55): 335-47.
- Bosmann M, Haggadone MD, Hemmila MR, et al. Complement activation product C5a is a selective suppressor of TLR4-induced, but not TLR3-induced, production of IL-27 (p28) from macrophages. Journal of Immunology 2012; 188 (10): 5086-93.
- Ding A, Hongfeng J, Westerterp M, et al. Disruption of mTORC1 in Macrophages Decreases Chemokine Gene Expression and Atherosclerosis. Circulation Research 2014; 114 (10): 1576-84.
- Naugler WE, Karin M. The wolf in sheep's clothing: the role of interleukin-6 in immunity, inflammation and cancer. Trends in Molecular Medicine 2008; 14 (3): 109-19.
- Goncharova EA, Goncharov DA, Damera G, et al. Signal transducer and activator of transcription 3 is required for abnormal proliferation and survival of tsc2-deficient cells: Relevance to pulmonary lymphangioleiomyomatosis. Molecular Pharmacology 2009 (76): 766-77.
- Schroer N, Pahne J, Walch B, et al. Molecular pathobiology of human cervical high-grade lesions: Paracrine stat3 activation in tumor-instructed myeloid cells drives local mmp-9 expression. Cancer Research 2011; (71): 87-97.
- Dyken van SJ, Locksley RM. Interleukin-4- and interleukin-13-mediated alternatively activated macrophages: roles in homeostasis and disease. Annu Rev Immunol 2013; 31: 317-43.
- Hudson CC, Liu M, Chiang GG, et al. Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Molecular Cell Biology 2002; 22 (20): 7004-14.
- Kim SY, Choi YJ, Joung SM, et al. Hypoxic stress up-regulates the expression of Toll-like receptor 4 in macrophages via hypoxia-inducible factor. Immunology 2010; 129 (4): 516-24.
- Wenger RH. Cellular adaptation to hypoxia: O2-sensing protein hydroxylases, hypoxia-inducible transcription factors, and O2-regulated gene expression. The FASEB Journal 2002; 16 (10): 1151-62.
- Semenza GL. Targeting hif-1 for cancer therapy. National Reviews. Cancer 2003; (3): 721-32.
- Csibi A, et al. The mTORC1/S6K1 pathway regulates glutamine metabolism through the eIF4B-dependent control of c-Myc translation. Current biology 2014; 24 (19): 2274-80.
- Sears R, Nuckolls F, Haura E, et al. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes & Development 2000; 14 (19): 2501-14.
- Martinez FO, Gordon S, Locati M, Mantovani A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. Journal of Immunology 2006; 177 (10): 7303-11.
- Xu X, Grijalva A, Skowronski A, et al. Obesity activates a program of lysosomal-dependent lipid metabolism in adipose tissue macrophages independently of classic activation. Cell Metabolism 2013; (18): 816-30.
- Liu H, Ma Y, Cole SM, et al. Serine phosphorylation of STAT3 is essential for Mcl-1 expression and macrophage survival. Blood 2003; 102 (1): 344-52.
- Philip B, Ito K, Moreno-Sánchez R, et al. HIF expression and the role of hypoxic microenvironments within primary tumors as protective sites driving cancer stem cell renewal and metastatic progression. Carcinogenesis 2013; 34 (8): 1699-707.
- Muraoka-Cook RS, Kurokawa H, Koh Y, et al. Conditional overexpression of active transforming growth factor beta1 in vivo accelerates metastases of transgenic mammary tumors. Cancer Research 2004; 64 (24): 9002-11.
- Chen W, Ma T, Shen X, et al. Macrophage-Induced Tumor Angiogenesis Is Regulated by the TSC2 — mTOR Pathway. Cancer Research 2012; (72): 1363.
- Ma J, Meng Y, Kwiatkowski DJ, et al. Mammalian target of rapamycin regulates murine and human cell differentiation
- through STAT3/p63/Jagged/Notch cascade. The Journal of Clinical Investigation 2010; (120): 103-14.
- Sumimoto H, Imabayashi F, Iwata T, et al. The BRAF-MAPK signaling pathway is essential for cancer-immune evasion in human melanoma cells. The Journal of Experimental Medicine 2006; (203): 1651-6.
- Cannon MJ, Ghosh D, Gujja S. Signaling Circuits and Regulation of Immune Suppression by Ovarian Tumor-Associated Macrophages. Vaccines (Basel) 2015; 3 (2): 448-66.
- Bosch van den MW M, Palsson-Mcdermott E, Johnson DS, et al. LPS Induces the Degradation of Programmed Cell Death Protein 4 (PDCD4) to Release Twist2, activating c-Maf Transcription to Promote Interleukin-10 Production. The Journal of Biological Chemistry 2014; (289): 22980-90.
- Kujawski M, Kortylewski M, Lee H, et al. Stat3 mediates myeloid cell-dependent tumor angiogenesis in mice. The Journal of Clinical Investigation 2008; 118 (10): 3367-77.
- Foekens JA, Peters HA, Look, et al. The urokinase system of plasminogen activation and prognosis in 2780 breast cancer patients. Cancer Res 2000; (60): 636-43.
- Bonde AK, Tischler V, Kumar S. Intratumoral macrophages contribute to epithelial-mesenchymal transition in solid tumors. BMC Cancer 2012; (12): 35.
- Thomas DA, Massague J. TGF-ß directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 2005; (8): 369-80.
- Sica A, Melillo G, Varesio L. Hypoxia: A double-edged sword of immunity. J Mol Med 2011; (89): 657-65.
