Роль натриевых каналов в механизме развития оксидативного стресса в модели ишемии/ реперфузии

Автор: Юрова Елена Валерьевна, Погодина Евгения Сергеевна, Расторгуева Евгения Владимировна, Белобородов Евгений Алексеевич, Сугак Дмитрий Евгеньевич, Фомин Александр Николаевич, Саенко Юрий Владимирович
Журнал: Ульяновский медико-биологический журнал @medbio-ulsu
Рубрика: Биологические науки
Статья в выпуске: 1, 2023 года.
Бесплатный доступ
Ишемическое и реперфузионное повреждение является критическим состоянием, при котором необходимо контролировать гибель клеток и сохранять функцию тканей. Восстановление потока питательных веществ и кислорода вызывает вторичное повреждение ишемизированных клеток и называется реперфузионным повреждением. Поскольку при развитии реперфузионного повреждения происходит, с одной стороны, колебание концентрации ионов внутри клеток, в частности ионов натрия, за счет изменения проводимости потенциалзависимых ионных каналов, а с другой -активация антиоксидантной системы как ответная реакция на оксидативный стресс, в которой ключевая роль отводится активным формам кислорода и оксиду азота II, особый интерес представляет влияние ингибиторов ионных каналов на механизмы развития оксидативного стресса, а также на апоптоз и некроз при реперфузии. Цель исследования. Изучение роли натриевых каналов в развитии оксидативного стресса при развитии ишемического и реперфузионного повреждения и при действии ингибитора натриевых каналов. Материалы и методы. Изучалось влияние синтезированного пептидного токсина ингибитора потенциалзависимых натриевых каналов при моделировании условий ишемии/реперфузии в культуре CHO-K1 на уровень апоптоза, некроза и оксидативного стресса (концентрация активных форм кислорода, оксида азота и глутатиона) с использованием флуоресцентных красителей и планшетного ридера-флуориметра. Результаты. Получены данные, указывающие на снижение уровня апоптоза и некроза, а также на поддержание концентрации оксида азота на контрольном уровне при добавлении токсина в нано-молярной концентрации. При этом концентрации активных форм кислорода и глутатиона не менялись. Таким образом, токсин-ингибитор проявил себя как защитный агент за счет предотвращения снижения концентрации оксида азота, что благоприятно сказалось на выживании клеточной культуры при реперфузии после ишемии.
Натрий, ишемия, реперфузия, токсин, ионные каналы, апоптоз
Короткий адрес: https://sciup.org/14127220
IDR: 14127220 | УДК: 576.36 | DOI: 10.34014/2227-1848-2023-1-145-154
Role of sodium channels in the development of oxidative stress in ischemia/reperfusion model
Ischemic and reperfusion injury is a critical condition, as it is necessary to control cell death and maintain tissue function. Restoration of nutrient and oxygen flow causes secondary damage to ischemic cells and is called reperfusion injury. Reperfusion injury causes, on the one hand, fluctuations in ion concentration inside cells, in particular sodium ions, due to changes in the conductivity of voltage-dependent ion channels, and, on the other hand, activation of the antioxidant system as a response to oxidative stress, in which the key role is given to reactive oxygen species and nitric oxide. Thus, the effect of ion channel inhibitors on the progression of oxidative stress, apoptosis and necrosis during reperfusion is of particular interest. The aim of the study is to examine the impact of sodium channels on oxidative stress under ischemic and reperfusion injury and sodium channel blockers action. Materials and Methods. The authors studied the influence of the synthesized peptide toxin, an inhibitor of voltage-gated sodium channels, under modelled ischemia/reperfusion in CHO-K1 culture on the level of apoptosis, necrosis, and oxidative stress (concentration of reactive oxygen species, nitric oxide, and glutathione) using fluorescent dyes and fluorescence microplate reader. Results. Data obtained indicate a decreased level of apoptosis and necrosis, and a control level of nitric oxide under toxin at a nanomolar concentration. At the same time, the concentrations of reactive oxygen species and glutathione did not change. Thus, the inhibitor toxin acted as a protective agent by preventing a decrease in the nitric oxide concentration, which favorably affected the survival of the cell culture during reperfusion after ischemia.
Текст научной статьи Роль натриевых каналов в механизме развития оксидативного стресса в модели ишемии/ реперфузии
Введение. Ишемическое и реперфузионное повреждение является критическим состоянием, при котором необходимо контролировать гибель клеток и сохранять функцию тканей. Ишемия определяется как гипоперфузия тканей. Некоторые состояния, такие как сепсис, острый коронарный синдром, а также трансплантация органов и травмы конечностей могут вызывать гипоперфузию тканей. Исследования показывают, что реперфузия приводит к последующему повреждению ишемизированной ткани, которое называется реперфузионным повреждением [1].
Ишемия может быть замаскирована под вполне терпимое явление, однако она работает как триггер для производства молекул, необходимых для индукции реперфузионного повреждения. При недостатке как кислорода, так и питательных веществ происходит отказ натрий-калиевых (насосы Na/K-АТФазы) и кальциевых насосов (насосы Са-АТФазы) на клеточной поверхности. Выход из строя насосов Na/K-АТФазы вызывает задержку натрия в клетках и калия вне клеток. Более высокий уровень натрия в клетках снижает активность натрий-водородных обменных насосов (Na/H-насосы). Кальциевые насосы в эндоплазматическом ретикулуме также перестают функционировать, что ограничивает обратный захват кальция. Накопление ионов водорода, натрия и кальция в клетках вызывает гиперосмолярность, что приводит к поступлению воды в ци- топлазму и набуханию клеток. Удержание водорода снижает клеточный pH, что приводит к нарушению активности ферментов и скоплению ядерного хроматина [2, 3]. При реперфузии активность обменных Na/H-насосов ускоряется за счет вымывания внеклеточных ионов H+, которые накапливались во время ишемии, что увеличивает протонный градиент через плазмолемму и дополнительно увеличивает цитозольный Ca2+. В попытке справиться с огромными изменениями уровней цитозольного Ca2+ увеличивается транспорт через митохондриальный унипортер Ca2+. Используя отрицательный митохондриальный потенциал, этот транспортер перемещает положительно заряженные ионы Ca2+ в митохондриальный матрикс [4, 5]. С одной стороны, происходит снижение цитозольного Са2+, с другой – повышение митохондриального Са2+, что вызывает открытие митохондриальной поры переходного типа, выделение цитохрома и запуск программ гибели клеток [6]. Механизмы реперфузионного повреждения включают оба типа гибели клеток – некротическую и апоптозную [3]. Периоды ишемии связаны с увеличением скорости некроза, тогда как реперфузия, как это ни парадоксально, приводит к усилению апоптоза. При реперфузии обеспечиваются необходимые условия для выживания жизнеспособных клеток, однако восстанавливается энергия, необходимая для завершения апоптоза [7, 8].
Приток кислорода, возникающий на стадии реперфузии, подпитывает избыточное производство активных форм кислорода (АФК), создавая парадокс, заключающийся в том, что эти высокореактивные частицы могут модифицировать белки, липиды, нуклеиновые кислоты, приводя к гибели клеток. В дополнение к этому в настоящее время признано, что окислительно-восстановительные молекулы, полученные из оксида азота II (NO), также вносят свой вклад в ишемическое и реперфузионное повреждение посредством окислительных и нитрозативных реакций практически с каждой биомолекулой, обнаруженной в клетках [9, 10]. Однако как NO, так и АФК в зависимости от их концентрации могут либо непосредственно участвовать в повреждении, либо обеспечивать защиту [11].
Таким образом, поскольку при развитии реперфузионного повреждения, с одной стороны, происходит изменение концентрации ионов внутри клеток, одна из стратегий снижения массовой гибели клеток связана с регуляцией концентрации ионов. Однако в большинстве исследований ведущая роль отводится ионам кальция как вторичным мессенджерам [12]. Поскольку ионы натрия также принимают активное участие в развитии механизмов гибели [13], в рамках нашего исследования акцент был сделан на ионы натрия и потенциалзависимые ионные каналы как основной путь проникновения ионов в клетку и изучалось влияние пептидного ингибитора натриевых каналов при реперфузии. В качестве ингибитора использовался токсин из семейства кноттинов, которые характеризуются наличием ингибиторного цистинового узла, придающего стабильность молекуле [14, 15]. Подобные токсины способны избирательно связываться с ионными каналами и ингибировать их действие.
С другой стороны, при развитии реперфузионного повреждения после ишемии ключевая роль отводится АФК и NO, а также активации антиоксидантной системы как ответной реакции на оксидативный стресс.
Цель исследования. Изучение роли натриевых каналов в развитии оксидативного стресса как при развитии ишемического и реперфузионного повреждения, так и при действии ингибитора натриевых каналов.
Материалы и методы. В работе было изучено влияние пептидного токсина – ингибитора натриевых потенциалзависимых каналов μ-agatoxin-Aa1a (UniProt: P11057). Токсин был синтезирован с использованием автоматического пептидного синтезатора ResPep SL (Intavis, Германия) по стандартному протоколу. Анализ токсина проводился методом обращенно-фазовой хроматографии с использованием хроматографической системы Shimadzu LC-20AD XR и колонки Dr. Maisch Luna C18(2) по стандартному протоколу градиентного элюирования от 95 % A, 5 % B до 0 % A, 100 % B в течение 40 мин, где элюент A – деионизированная вода, элюент B – ацетонитрил. Детектирование осуществлялось на длине волны 215 нм. Масс-спектрометрический анализ проводился с использованием программно-аппаратного комплекса MALDI-TOF MS FLEX series (Bruker Daltonics, Германия). Очистка производилась с помощью высокоэффективной жидкостной хроматографии с использованием системы хроматографии NGC Quest™ 10 (Bio-Rad, США) на колонке Econo-Column 1x30cm (Bio-Rad, США).
Исследование было проведено на культуре CHO-K1 эпителиального происхождения. Культура содержалась при 37 °С и 5 % СО 2 в среде DMEM/F12 («ПанЭко», Россия) с 10 % фетальной бычьей сывороткой (Biosera, Франция) и гентамицином, пассажи делались каждые 3–4 дня. Для воспроизведений ишемического и реперфузионного повреждения культура на экспоненциальной стадии роста помещалась на 3 ч в условия ишемии (среда DMEM с пониженным содержанием глюкозы (1 г/л), сыворотки (1 %) и кислорода (1 %)), затем на 3 ч в условия реперфузии (DMEM с содержанием глюкозы 3,15 г/л, сыворотки 10 % и кислорода 18,6 %) с токсином в концентрации
50 нМ и без него. По завершении реперфузии фиксировался уровень апоптоза, некроза, концентрация АФК, NO и глутатиона. Измерение проводилось с использованием планшетного ридера-флуориметра CLARIO star Plus (BMG LABTECH, Германия) и флуоресцентных красителей: Yo-PRO 1 PI (1 мкМ) для апоптоза и некроза, DCFH DA (1 мкМ) для АФК, DAF FM (1 мкМ) для NO, MCB (1 мкМ) для глутатиона. Первичная обработка данных проводилась с использованием ПО Mars (BMG LABTECH, Германия). Данные были пересчитаны на 100 000 клеток. Статистическая обработка данных осуществлялась с использованием критерия Манна – Уитни.
Результаты. В результате исследования было выявлено, что уровень апоптоза при моделировании условий ишемии/реперфузии повышается относительно контрольных условий. Однако при наличии в среде для реперфузии токсина в концентрации 50 нМ апоптоз остается на уровне нормальных условий (рис. 1).
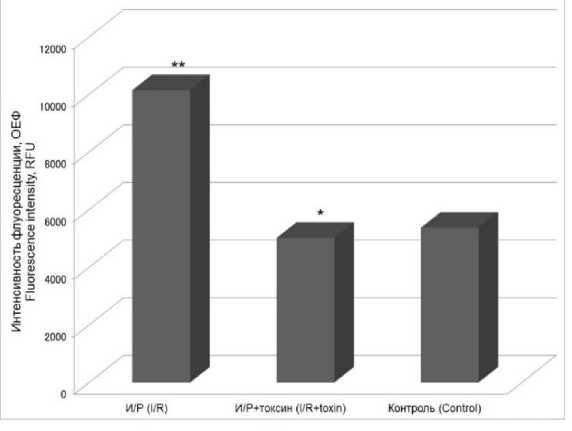
Рис. 1. Изменение уровня апоптоза в культуре CHO-K1 при действии токсина в концентрации 50 нМ в условиях ишемии/реперфузии (И/Р) (ОЕФ – относительные единицы флуоресценции;
* – p<0,05 при сравнении с экспериментальной группой (И/Р);
** – p<0,05 при сравнении с контролем. Далее обозначения те же)
Fig. 1. Change in the apoptosis level in CHO-K1 culture under ischemia/reperfusion (I/R), toxin concentration 50 nM (RFU – relative fluorescent unit;
* – p<0.05 the differences are significant when compared to the experimental group (I/R);
** – p<0.05 the differences are significant when compared to the control group)
Далее было обнаружено, что аналогично апоптозу уровень некроза в условиях ише-мии/реперфузии возрастает. Добавление ток- сина в концентрации 50 нМ также поддерживает уровень некроза на уровне контрольных условий (рис. 2).
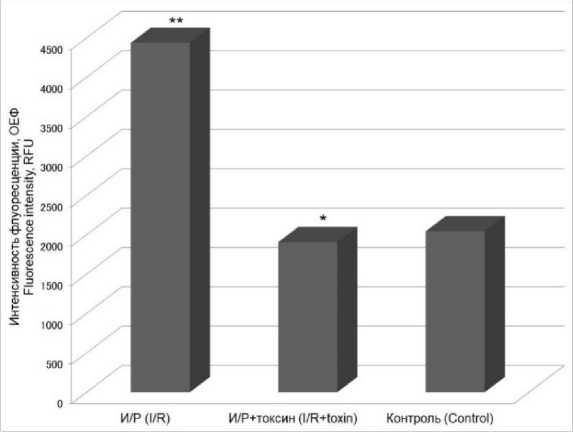
Рис. 2. Изменение уровня некроза в культуре CHO-K1 при действии токсина в концентрации 50 нМ в условиях ишемии/реперфузии
Fig. 2. Change in the necrosis level in CHO-K1 culture under ischemia/reperfusion (I/R), toxin concentration 50 nM (RFU – relative fluorescent unit;
* – p<0.05 the differences are significant when compared to the experimental group (I/R);
** – p<0.05 the differences are significant when compared to the control group)
На рис. 3, где представлено изменение концентрации активных форм кислорода при моделировании условий ишемии/реперфузии в присутствии токсина и без него, видно, что токсин дополнительно снижает внутриклеточ- ную концентрацию АФК относительно контрольных условий, несмотря на то что при реперфузии концентрация АФК в наших экспериментах не повышается.
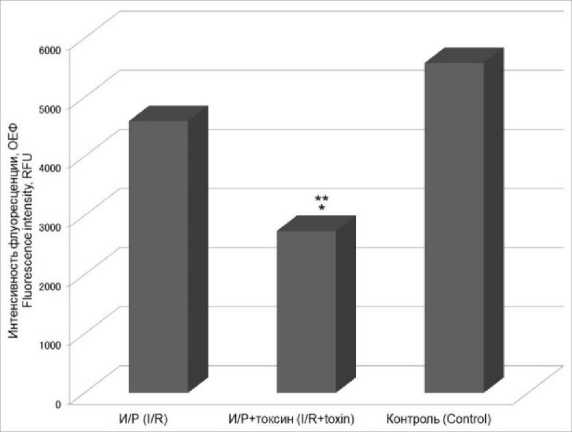
Рис. 3. Изменение концентрации активных форм кислорода в культуре CHO-K1 при действии токсина в концентрации 50 нМ в условиях ишемии/реперфузии
Fig. 3. Change in the concentration of reactive oxygen species in CHO-K1 culture under ischemia/reperfusion (I/R), toxin concentration 50 nM (RFU – relative fluorescent unit;
* – p<0.05 the differences are significant when compared to the experimental group (I/R);
** – p<0.05 the differences are significant when compared to the control group)
При моделировании условий ишемии/ре-перфузии изменения концентрации глутатиона не происходит как при реперфузии без токсина, так и при реперфузии в присутствии токсина (рис. 4).
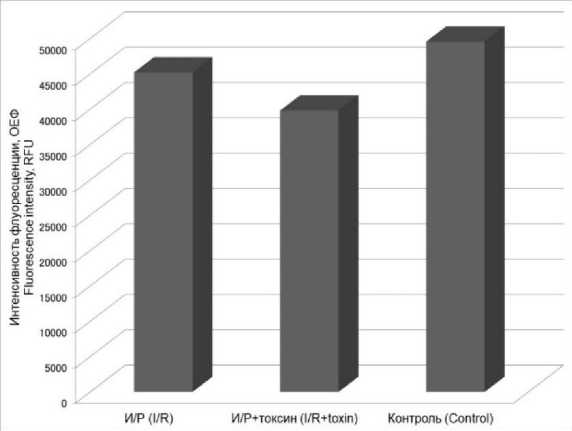
Рис. 4. Изменение концентрации глутатиона в культуре CHO-K1 при действии токсина в концентрации 50 нМ в условиях ишемии/реперфузии
Fig. 4. Change in glutathione concentration in CHO-K1 culture under ischemia/reperfusion (I/R), toxin concentration 50 nM (RFU – relative fluorescent unit;
-
* – p<0.05 the differences are significant when compared to the experimental group (I/R);
** – p<0.05 the differences are significant when compared to the control group)
На рис. 5 видно, что при ишемии/репер-фузии в культуре происходит снижение концентрации оксида азота ниже контрольного уровня. Однако добавление токсина поддерживает концентрацию оксида азота на контрольном уровне.
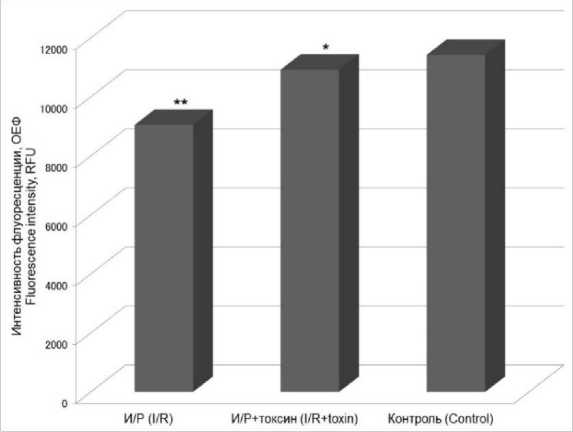
Рис. 5. Изменение концентрации оксида азота (II) в культуре CHO-K1 при действии токсина в концентрации 50 нМ в условиях ишемии/реперфузии
Fig. 5. Change in nitric oxide concentration in CHO-K1 culture under ischemia/reperfusion (I/R), toxin concentration 50 nM (RFU – relative fluorescent unit;
-
* – p<0.05 the differences are significant when compared to the experimental group (I/R);
** – p<0.05 the differences are significant when compared to the control group)
Обсуждение. В наших экспериментах было изучено влияние токсина-ингибитора натриевых потенциалзависимых каналов на апоптоз, некроз и оксидативный стресс клеточной культуры CHO-K1 при моделировании условий ишемии/реперфузии. Апоптоз как запрограммированная гибель клеток тонко регулируется на генном уровне, что приводит к упорядоченному и эффективному удалению поврежденных клеток. Механизм апоптоза сложен и включает множество сигнальных путей [16]. В то время как некроз, также известный как незапрограммированная гибель клеток, вызывает морфологические изменения, включая набухание клеток или ядер и разрушение плазматической мембраны [17]. Для развития ишемического и реперфузионного повреждения характерны оба типа гибели клеток. Общее повреждение тканей, вызванное ише-мией/реперфузией, делится на две части: ишемическое повреждение (истощение клеточной энергии) и реперфузионное повреждение (резкое восстановление потока питательных веществ и кислорода). После ишемии продукты обмена веществ задерживаются в клетке и вызывают ацидоз. На стадии реперфузии продукция активных форм кислорода усиливается, что приводит к вторичному повреждению.
Данные наших экспериментов указывают на развитие и апоптоза, и некроза при моделировании условий ишемии/реперфузии в клеточной культуре (рис. 1, 2). Добавление токсина в концентрации 50 нМ носит защитный характер и предотвращает массовую гибель клеток. Как упоминалось выше, развитие как апоптоза, так и некроза при реперфузии связано с повышением АФК. Однако в наших экспериментах подобного не отмечается (рис. 3), что также коррелирует с данными об изменении концентрации глутатиона (рис. 4), который должен выступать в роли антиоксидантной системы. Один из вероятных механизмов подобного действия связан с использованием эпителиальной клеточной культуры. Несмотря на то что данная культура подвержена ишемическому и реперфузионному повреждению, которое проявляется в виде развития апоптоза и некроза, механизм повреждения носит иной характер. Чувствительность клеток к гипоксическому воздействию варьирует среди различных типов тканей. Хотя существуют различные способы запуска апоптоза и некроза, морфологические паттерны остаются неизменными. Следовательно, каким бы ни был стимул, существует общий конечный путь, который может привести к гибели клеток. Однако стоит отметить, что токсин в данном случае снижает физиологическую концентрацию активных форм кислорода, что никак не сказывается на изменении других параметров и не оказывает влияния на концентрацию глутатиона. С другой стороны, параллельный оксида-тивному стрессу процесс связан с изменением концентрации оксида азота. Многочисленные эксперименты свидетельствуют о том, что NO снижает неблагоприятный эффект ишемии/ реперфузии [18]. У мышей с нокаутом эндотелиальной синтазы оксида азота (NOS3) функциональное восстановление после ишемии/ре-перфузии ослаблено [19], в то время как увеличение концентрации NO ускоряет функциональное восстановление клеток [20]. В нашем случае при моделировании условий ише-мии/реперфузии происходит снижение концентрации оксида азота, что коррелирует с увеличением апоптоза и некроза. С другой стороны, добавление токсина препятствует снижению концентрации NO (рис. 5), что в свою очередь благоприятно сказывается на выживании клеток (рис. 1, 2).
Заключение. Таким образом, проведенное исследование продемонстрировало, что пептидный токсин – ингибитор натриевых потенциалзависимых каналов предотвращает массовую гибель клеток в культуре CHO-K1 при моделировании условий ишемии/репер-фузии за счет ингибирования развития апоптоза и некроза. Однако, хотя изучаемая культура и подвергается ишемическому и реперфузионному повреждению, механизм гибели не связан с развитием оксидативного стресса. Несмотря на это, изучаемый токсин может рассматриваться в качестве защитной стратегии для предотвращения массовой гибели клеток в клеточной модели ишемии/ре-перфузии.
Работа выполнена при финансовой поддержке Министерства науки и высшего образования Российской Федерации (проект № FEUF-2022-0008).
Список литературы Роль натриевых каналов в механизме развития оксидативного стресса в модели ишемии/ реперфузии
- НеймаркМ.И. Синдром ишемии-реперфузии. Хирургия. Журнал им. Н.И. Пирогова. 2021; 9: 71-76.
- Wu M.Y., Yiang G.T., Liao W.T., Tsai A.P., Cheng Y.L., Cheng P. W., Li C.Y., Li C.J. Current Mechanistic Concepts in Ischemia and Reperfusion Injury. Cell Physiol Biochem. 2018; 46 (4): 1650-1667.
- Lopez-Neblina F., Toledo A.H., Toledo-Pereyra L.H. Molecular biology of apoptosis in ischemia and reperfusion. J Invest Surg. 2005; 18 (6): 335-350.
- Contreras L., Drago I., Zampese E., Pozzan T. Mitochondria: the calcium connection. Biochim Biophys Acta. 2010; 1797 (6-7): 607-618.
- SzydlowskaK., TymianskiM. Calcium, ischemia and excitotoxicity. Cell Calcium. 2010; 47 (2): 122-129.
- Kalogeris T., Baines C.P., Krenz M., Korthuis R.J. Ischemia/Reperfusion. Compr Physiol. 2016; 7 (1): 113-170.
- Soares R.O.S., Losada D.M., Jordani M.C., Évora P., Castro-E-Silva O. Ischemia/Reperfusion Injury Revisited: An Overview of the Latest Pharmacological Strategies. Int J Mol Sci. 2019; 20 (20): 5034.
- Eefting F., Rensing B., Wigman J., Pannekoek W.J., Liu W.M., Cramer M.J., Lips D.J., Doevendans P.A. Role of apoptosis in reperfusion injury. Cardiovasc Res. 2004; 61 (3): 414-426.
- Duan J., Kasper D.L. Oxidative depolymerization of polysaccharides by reactive oxygen/nitrogen species. Glycobiology. 2011; 21 (4): 401-409.
- Kvietys P.R., Granger D.N. Role of reactive oxygen and nitrogen species in the vascular responses to inflammation. Free Radic Biol Med. 2012; 52 (3): 556-592.
- Folino A., Losano G., Rastaldo R. Balance of nitric oxide and reactive oxygen species in myocardial reperfusion injury and protection. J Cardiovasc Pharmacol. 2013; 62 (6): 567-575.
- Saber M., Eimani H., Soleimani Mehranjani M., Shahverdi A., Momeni H.R., Fathi R., Tavana S. The effect of Verapamil on ischaemia/reperfusion injury in mouse ovarian tissue transplantation. Biomed Pharmacother. 2018; 108: 1313-1319.
- Kondratskyi A., Kondratska K., Skryma R., Prevarskaya N. Ion channels in the regulation of apoptosis. Biochim Biophys Acta. 2015; 1848 (10, Pt. B): 2532-2546.
- Василевский А.А., Козлов С.А., Гришин Е.В. Молекулярное разнообразие яда пауков. Успехи биологической химии. 2009; 49: 211-274.
- Kalia J., Milescu M., Salvatierra J., Wagner J., Klint J.K., King G.F., Olivera B.M., Bosmans F. From foe to friend: using animal toxins to investigate ion channel function. J Mol Biol. 2015; 427 (1): 158-175.
- Матвеева Н.Ю. Апоптоз: морфологические особенности и молекулярные механизмы. Тихоокеанский медицинский журнал. 2003; 4: 12-16.
- Mierke C.T. Cell Proliferation, Survival, Necrosis and Apoptosis. Springer: Cham; 2020: 743-824.
- Schulz R., Kelm M., Heusch G. Nitric oxide in myocardial ischemia/reperfusion injury. Cardiovasc Res. 2004; 61 (3): 402-413.
- Jones S.P., Girod W.G., Palazzo A.J., Granger D.N., Grisham M.B., JourdHeuil D., Huang P.L., Lefer D.J. Myocardial ischemia-reperfusion injury is exacerbated in absence of endothelial cell nitric oxide synthase. Am J Physiol. 1999; 276 (5): 1567-1573.
- Kanno S., Lee P.C., Zhang Y., Ho C., Griffith B.P., ShearsL.L. 2nd, Billiar T.R. Attenuation of myocardial ischemia/reperfusion injury by superinduction of inducible nitric oxide synthase. Circulation. 2000; 101 (23): 2742-1748.
