Стратегия разработки таргетных препаратов для терапии HER2-позитивного рака молочной железы

Автор: Боденко В.В., Ларькина М.С., Третьякова М.С., Белоусов М.В., Чернов В.И.
Журнал: Сибирский онкологический журнал @siboncoj
Рубрика: Обзоры
Статья в выпуске: 3 т.24, 2025 года.
Бесплатный доступ
Цель исследования – определить современные стратегии, достижения и проблемы в разработке таргетных HER2-направленных терапевтических конъюгатов и текущие клинические результаты терапии на их основе. Материал и методы. Изучено 247 литературных источников в пределах 7 лет, из которых для обзора отобрано 60. Поиск источников проведен в информационных базах данных Scopus (n=35), PubMed (n=121), WOS (n=91). Для получения полнотекстовых документов использованы электронные ресурсы PubMed Central (PMC), Science Direct, Research Gate. Результаты. Перспективность применения терапевтических HER2-направленных конъюгатов клинически подтверждена для таких препаратов, как трастузумаб эмтанзин (T-DM1, Kadcyla®), трастузумаб дерукстекан (T-DXd, Enhertu®). Разработка новых HER2-направленных конъюгатов включает ряд подходов, направленных на улучшение характеристик препаратов и повышение эффективности терапии: увеличение аффинности связывания с мишенью, одновременное блокирование двух доменов HER2, различные механизмы цитотоксического воздействия на опухолевую клетку (ингибирование тубулина, РНК-полимеразы II, ДНКтопоизомеразы I, повреждение ДНК), увеличение соотношения цитотоксических агентов на молекулу антитела, использование «эффекта свидетеля», повышение стабильности конъюгата в кровотоке. На стадии клинических исследований изучаются такие препараты, как трастузумаб дуокармазин (SYD985), Диситамаб ведотин (RC48, Aidixi®), Занидатамаб зоводотин (ZW49), ARX788, MRG002. Заключение. Совершенствование конструкции и понимание нюансов взаимодействия таргетных конъюгатов и опухоли способствуют новым клиническим успехам, что может обеспечить не только преимущество в выживании по сравнению с традиционной терапией, но и значительно улучшить качество жизни пациентов с HER2-положительным статусом.
Рак молочной железы, анти-HER2 терапия, таргетный конъюгат, цитотоксический агент, трастузумаб эмтанзин, трастузумаб дерукстекан, трастузумаб дуокармазин
Короткий адрес: https://sciup.org/140310581
IDR: 140310581 | УДК: 618.19-006.6:615.277.3:615.065 | DOI: 10.21294/1814-4861-2025-24-3-135-148
Strategy for the development of targeted agents for therapy of HER2-positive breast cancer
The objective of the study: to identify current strategies, achievements, and challenges in developing HER2-targeted therapeutic conjugates and assess clinical outcomes of therapies based on them. Material and Methods. A total of 247 publications over the past 7 years were analyzed. Finally, 60 were selected for the review. The sources were searched in Scopus (n=35), PubMed (n=321) and WOS (n=91) databases. Electronic resources, such as PubMed Central (PMC), ScienceDirect and ResearchGate were used to obtain the full-text articles. Results. The therapeutic potential of HER2-targeted conjugates has been confirmed in clinical trials for drugs, such as trastuzumab emtansine (T-DM1, Kadcyla®) and trastuzumab deruxtecan (T-DXd, Enhertu®). The development of new HER2-targeted conjugates involves several approaches to improve drug characteristics and therapeutic efficacy: increasing binding affinity to the target, simultaneous blocking of two HER2 domains, various mechanisms of cytotoxic effects on tumor cells (inhibition of tubulin, RNA polymerase II, and DNA topoisomerase I), enhancing the ratio of cytotoxic agents per antibody molecule and improving the conjugate stability in the bloodstream. Trastuzumab duocarmazine (SYD985), disitamab vedotin (RC48, Aidixi®), zanidatamab zovodotin (ZW49), ARX788 and MRG002 are all currently being studied in clinical trials. Conclusion. Improvements in the design and understanding of drug-tumor interactions contribute to new clinical advances that may provide not only a survival advantage over traditional therapy, but also significantly improve the quality of life of patients with HER2-positive tumors.
Текст научной статьи Стратегия разработки таргетных препаратов для терапии HER2-позитивного рака молочной железы
Рак молочной железы (РМЖ) представляет собой гетерогенное заболевание, включающее в себя молекулярные подтипы c различными гистологическими моделями, паттернами развития и прогрессирования. Активация мутаций HER2 является ранним событием онкогенеза молочной железы, приводящим к гиперэкспрессии HER2 у 20–25 % больных РМЖ. В результате амплификации гена HER2/neu или нарушения регуляции транскрипции РМЖ может иметь до 50 копий гена HER2 и достигать 100-кратного увеличения экспрессии белка HER2, что приводит к появлению 2–10 млн рецепторов, экспрессируемых на поверхности опухолевых клеток [1]. При этом гиперэкспрессия HER2, в дополнение к усилению передачи сигналов посредством лиганд-зависимой гетеродимеризации, приводит к лиганд-независимой димеризации рецептора и аномальной передаче сигналов. В результате аномальных молекулярных взаимодействий HER2-положительный статус способствует развитию агрессивных фенотипов заболеваний, сокращению общей выживаемости пациентов, а также устойчивости к химиотерапии, гормональной и лучевой терапии. HER2-положительный статус способствует метастазированию и повышает вероятность рецидива заболевания [2].
Клиническое значение диагностики HER2-положительного подтипа РМЖ изменилось с момента появления анти-HER2-таргетной терапии. Повышенная экспрессия HER2 при РМЖ несет важную прогностическую информацию, которая позволяет использовать таргетную анти-HER2-направленную терапию, способную улучшить результаты лечения. Наличие побочных эффектов, первичная и приобретенная резистентность являются основными ограничениями такого лечения. Еще одной проблемой, препятствующей применению анти-HER2-направленной терапии в монорежиме, является гетерогенность опухоли, когда клетки с гиперэкспрессией рецепторов HER2 соседствуют с HER2-отрицательными [3]. Вследствие этого сохраняется актуальность продолжения исследований, направленных на создание таргетных лекарственных препаратов для лечения опухолей с HER2-положительным статусом.
Стратегия разработки таргетных препаратов для терапии HER2-позитивных злокачественных новообразований
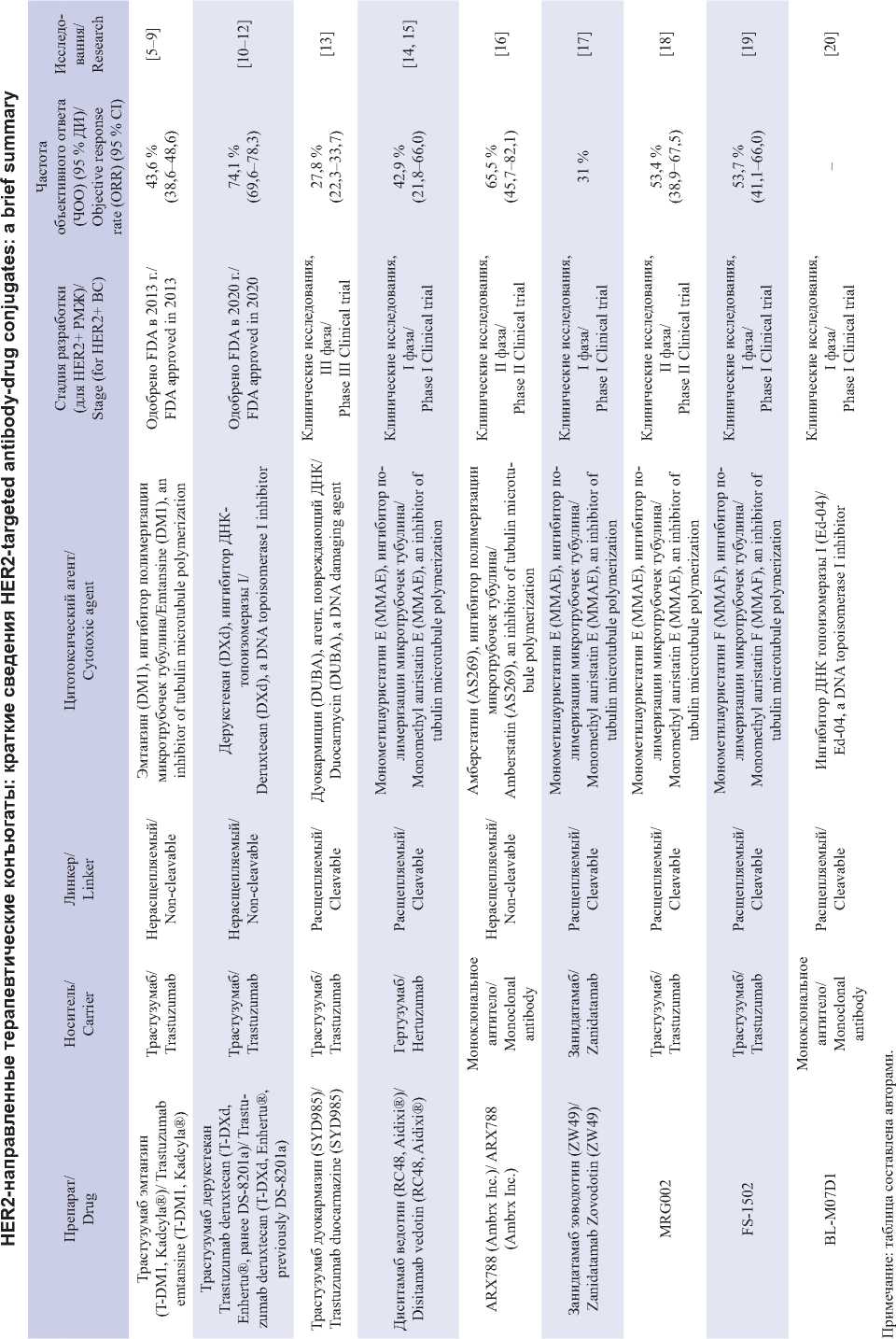
Технологическим прорывом в таргетной анти-HER2-терапии стали конъюгаты моноклональных антител (мкАТ) с цитотоксическими агентами (таблица), первым из которых стал трастузумаб эмтанзин. Если ранее в таргетной терапии целевая мишень рассматривалась только как биологический переключатель, включаемый или выключаемый с помощью антител и низкомолекулярных ингибиторов тирозинкиназы, то на данный момент мишень рассматривается в качестве якоря для стыковки и связывания с конъюгатами, несущими цитотоксический агент и способными не просто ингибировать нижестоящую передачу сигналов, но и приводить к гибели целевой опухолевой клетки, а для определенных препаратов и соседствующих опухолевых клеток. Перспективность данного подхода для HER2-положительного РМЖ клинически подтверждена для таких препаратов, как трастузумаб эмтанзин, трастузумаб дерукстекан [4].
Разработка новых HER2-направленных конъюгатов включает различные подходы, направленные на улучшение характеристик препаратов и повышение эффективности терапии: увеличение аффинности связывания с мишенью, одновременное блокирование двух доменов HER2, различные механизмы цитотоксического воздействия на опухолевую клетку, увеличение соотношения цитотоксических агентов на молекулу антитела, использование «эффекта свидетеля», повышение стабильности конъюгата в кровотоке. Данные подходы позволяют снижать токсические эффекты, являются способом воздействия на опухоли с развившейся резистентностью к лечению и расширяют спектр использования препаратов при различных уровнях экспрессии HER2 [4]. За последние 30 лет разработан достаточно широкий спектр новых линкеров, цитотоксических агентов и стратегий конъюгации [21]. К сожалению, немногие из них дошли до применения в клинической практике. Это демонстрирует степень сложности оптимизации ключевых параметров таргетных конъюгатов. В данном обзоре мы попытались представить существующие подходы, обсудить достижения и проблемы в разработке терапевтических конъюгатов и текущие клинические результаты терапии на их основе.
Структура конъюгатов
Конъюгаты антител с лекарственным средством представляют собой моноклональное антитело (мкАТ), ковалентно связанное с низкомолекулярным цитотоксическим агентом посредством синтетического линкера. Антитело в результате связывания с целевой мишенью на поверхности опухолевой клетки и последующей интернализации конъюгата высвобождает высокоэффективный цитотоксический агент внутрь опухолевой клетки, обеспечивая ее гибель. Гибель пролиферативных клеток опухоли обеспечивается посредством ряда механизмов, таких как блокирование сборки тубулиновых микротрубочек с последующей остановкой клеточного цикла в фазе G2/M (эм-танзин), повреждение ДНК на различных фазах клеточного цикла (дуокармазин), ингибирование РНК-полимеразы II (аманитин) или топоизомеразы I (дерукстекан, говитекан). Цитотоксические агенты с направленной доставкой обладают в 100–1000 раз более выраженным терапевтическим эффектом по сравнению со стандартной химиотерапией, сохраняя допустимый уровень токсичности. Выбор структурных компонентов и их правильное сочетание в одном препарате являются ключом к созданию успешного лекарственного средства [22].
Носитель
При разработке и выборе носителя для терапевтического конъюгата необходимо учитывать множество факторов, каждый из которых оказывает прямое влияние на конечные характеристики препарата. В случае правильного выбора специфично нацеленная молекула-носитель опосредует точность воздействия на целевые опухолевые клетки, увеличивает терапевтический индекс и минимизирует возможные нецелевые токсические эффекты. Первым важным аспектом является выбор подкласса и изотипа мкАТ. В составе одобренных к применению препаратов в качестве молекулы-носителя преимущественно используют терапевтические, полностью гуманизированные мкАТ изотипа IgG, в частности IgG1. Изотипы IgG2 и IgG4 менее эффективны в модуляции эффекторных функций по сравнению с IgG1. Изотипы IgG3 обладают сравнительно коротким периодом полувыведения (~7 дней), имеют расширенную шарнирную область по сравнению с другими изотипами и склонны к полиморфизму и иммуногенным реакциям [23].
Структура мкАТ содержит антигенсвязывающий фрагмент (Fab), специфично взаимодействующий с антигеном опухолевой клетки, и кристаллизующийся фрагмент (Fc). Связывание Fc-домена мкАТ с лейкоцитарными Fc-рецепторами (FcRn) запускает иммунные эффекторные функции: антителозависимую клеточную цитотоксичность (ADCC) и комплемент-зависимую цитотоксичность (CDC). Адаптивный иммунный ответ оказывает дополнительный, не зависимый от воздействия цитотоксического агента, терапевтический эффект для устранения злокачественных клеток [24]. Доказано, что ADCC значительно увеличивает терапевтическую активность трастузумаба [25].
Другим аспектом влияния Fc-фрагмента IgG является FcRn-опосредованная рециркуляция, значительно увеличивающая период полувыведения мкАТ по сравнению с другими белками плазмы крови (~21 день) [26]. Fc-фрагмент IgG связывается
Òàблицà /Table
Note: created by the authors.
с неонатальным рецептором FcRn рН-зависимым образом (в подкисленной среде эндосом, рН=~6,0). После интернализации IgG внутрь клетки процесс FcRn-опосредованной рециркуляции сохраняет эндосомальный IgG от деградации посредством связывания в просвете эндосомы и последующего высвобождения во внеклеточную среду [27].
Дальнейший выбор мкАТ для создания терапевтических конъюгатов основан как на аффинности их связывания с целевой мишенью (kd<10 нМ), экспрессируемой на поверхности опухолевой клетки, так и на способности проникать внутрь опухоли. Как обособленный показатель высокая аффинность связывания антитела с целевым антигеном не гарантирует высокое опухолевое поглощение. Было обнаружено, что мкАТ с высокой аффинностью локализуются в периваскулярных пространствах, тогда как мкАТ с более низким уровнем связывания могут более эффективно интернализовываться внутри опухолевой клетки [28]. Феномен объясняется формированием «барьера сайта связывания», повышение уровня которого коррелирует с увеличением плотности целевых антигенов и аффинности направленных макромолекул. Таким образом, только баланс между скоростями интернализации и диссоциации комплексов «антиген-антитело» может обеспечить эффективную доставку цитотоксического агента в опухолевую клетку. Важны также такие базисные фармакокинетические параметры выбранного носителя, как длительный период полувыведения и медленный клиренс из плазмы крови [28]. Данные показатели, как правило, обеспечиваются большим размером макромолекул мкАТ (~150 кДа) и механизмами FcRn-опосредованной рециркуляции.
Несмотря на многочисленные достоинства, у мкАТ, используемых в качестве носителя цитотоксической нагрузки, существует ряд недостатков – нелинейное распределение и выведение, а также иммуногенность. Поэтому в качестве носителя также рассматриваются фрагменты антител и пептидные молекулы [29]. Малый размер таких носителей способствует эффективному нацеливанию на мишень и проникновению в солидные опухоли, и они продемонстрировали успех в доклинических и клинических исследованиях в качестве транспортных тераностических молекул [30, 31]. Однако конъюгируемый цитотоксический агент, размер которого сопоставим с размером носителя, вносит большие коррективы в биохимические и биофизические характеристики соединения и существенно, зачастую неблагоприятно, влияет на способность проникать внутрь опухолевой клетки и дальнейшее биораспределение конъюгата [29]. Данное направление является перспективным для терапии, но требует новых разработок и скурпулезных исследований влияния структурных компонентов конъюгата на его свойства. Пример успешных доклинических исследований терапевтических молекул на основе полипептидов, таких как каркасные белки аффибоди, продемонстрировали V. Tolmachev et al. [32–35].
Линкер
Основные требования к линкеру в составе терапевтического конъюгата включают возможность обеспечения стабильности конструкции препарата в системном кровообращении в течение длительного периода и целевое высвобождение цитотоксического агента после интернализации в опухолевой клетке. По механизму высвобождения цитотоксической нагрузки линкеры подразделяются на два обширных класса: расщепляемые и нерасщепляемые [22].
Расщепляемые линкеры длительное время стабильны в кровообращении и высвобождают цитотоксический агент в микроокружении опухоли. К ним относят химически лабильные линкеры (гидролизующиеся в кислой среде эндосом и лизосом, чувствительные к глутатиону), а также линкеры, расщепляемые лизосомальными протеазами (пептидные, расщепляемые β-глюкуронидазой, β-галактозидазой или фосфатазой). К основному преимуществу расщепляемых линкеров относится «эффект свидетеля» – воздействие на соседние опухолевые клетки с низкой экспрессией целевой мишени. При этом химически лабильным линкерам свойственна ограниченная стабильность в плазме, что грозит преждевременным высвобождением цитотоксина. Альтернативная стратегия использования линкеров, расщепляемых ферментами клеточных компартментов, увеличивает потенциал контролируемого высвобождения химиотерапевтического агента.
Нерасщепляемые линкеры, представителями которых являются тиоэфиры или малеимидокапро-илы (mc), характеризуются стабильными связями. Для высвобождения цитотоксина таким линкерам необходима полная лизосомальная ферментативная деградация в результате интернализации терапевтического конъюгата. Преимуществами нерасщепляемых линкеров являются их высокая стабильность в плазме и возможность увеличения терапевтического окна с минимизацией нецелевых токсических эффектов [36].
Цитотоксический агент
Используемый химиотерапевтический агент должен отвечать комплексу требований, первым из которых является чрезвычайно высокая эффективность в пикомолярных или наномолярных концентрациях. Это требование связано с ограничением количества опухолеспецифических мишеней в новообразовании, особенно в солидных опухолях. Кроме того, ввиду невысокой проницаемости и плохой интернализации мкАТ количество молекул цитотоксических агентов, которые могут быть эндоцитозированы в опухолевые клетки, может быть небольшим. Другими немаловажными требованиями являются стабильность цитотоксического агента в крови, среде эндосом и лизосом с низким pH; наличие функциональной группы, предназначенной для связывания с носителем посредством линкера и доступной для модификаций без значительного влияния на эффективность; низкая иммуногенность [37]. Для реализации «эффекта свидетеля» перспективными являются низкомолекулярные цитотоксические молекулы, проницаемые для гидрофобных клеточных мембран.
Ингибиторы тубулина
Одними из часто используемых в терапевтических конъюгатах цитотоксинов являются агенты, воздействующие на структуру микротрубочек. Микротрубочки представляют собой динамические полимеры цилиндрической формы, состоящие из гетеродимеров α-, β-тубулина. После митоза происходит деполимеризация микротрубочек, составляющих веретено деления. Следовательно, при нарушении равновесия процессов полимеризации и деполимеризации тубулиновых микротрубочек митоз останавливается в фазе G2/M. Химиотерапевтические агенты, нацеленные на тубулин, нарушая фазу полимеризации микротрубочек и разрушая митотическое веретено, останавливают G2/M фазу клеточного цикла и приводят к митотической катастрофе с последующим апоптозом клетки. Как следствие, токсичность ингибиторов тубулина направлена преимущественно на высокопролиферативные опухолевые клетки. В зависимости от механизма действия среди антимитотических агентов выделяют промоторы полимеризации тубулина (стабилизаторы микротрубочек) и ингибиторы полимеризации тубулина (дестабилизаторы микротрубочек). Ауристатины, стабилизаторы микротрубочек, представляют собой водорастворимые синтетические аналоги морского продукта доластатина 10, полученного из моллюска Dolabella auricleria . Производные ауристатинов, такие как MMAE (монометил ау-ристатин E), MMAF (монометил ауристатин F), нарушают функционирование микротрубочек, воздействуя на β-субъединицу димеров тубулина и определяя их нерегулируемый рост. Производные майтанзина, дестабилизаторы микротрубочек, представляют собой класс бензоансамкролидных антибиотиков, первоначально были выделены из коры африканского кустарника Maytenus ovatus . Наиболее известными представителями на данный момент являются DM1 (эмтанзин) и DM4 (равтанзин), которые, напротив, блокируя полимеризацию димеров тубулина, дестабилизируют микротрубочки и нарушают образование зрелых микротрубочек [37, 38].
Цитотоксины, повреждающие ДНК
Другим широко применяемым классом химиотерапевтических агентов являются цитотоксины, повреждающие ДНК. Ингибиторы ДНК связыва- ются с малой бороздкой двойной спирали ДНК и разрушают ее посредством ряда механизмов: двуцепочечного разрыва (калихеамицины), алкилирования (пирролобензодиазепины и дуокармицины), интеркаляции (камптотецины и антрациклины) или перекрестного сшивания ДНК. Для соединений, повреждающих ДНК, выделяют два главных преимущества: эффективность механизма действия на протяжении всего клеточного цикла и высокую цитотоксичность. Данный класс обладает особо высокоактивными цитотоксическими свойствами со значениями IC50 в пикомолярном диапазоне, в отличие от преимущественно наномолярного диапазона для ингибиторов микротрубочек, и, следовательно, может быть более эффективным в отношении солидных опухолей, а также в клетках с низкой экспрессией целевого антигена [37]. Аналог дуокармицина DUBA (дуокармицин-гидроксибензамид-азаиндол) использован в нескольких терапевтических конъюгатах нового поколения, в частности в HER2-направленном трастузумабе дуокармазине (SYD985).
Ингибиторы РНК-полимеразы II
РНК-полимераза II – фермент, осуществляющий синтез мРНК и многих ядерных РНК, играет ключевую роль в экспрессии генов и клеточной регуляции. Ингибиторы РНК-полимеразы II напрямую блокируют транскрипцию ДНК в мРНК, что в конечном итоге приводит к более чем 1000-кратному снижению транскрипции и синтеза белка с последующим апоптозом как делящихся, так и покоящихся клеток [38, 39]. Амантины, ингибиторы РНК-полимеразы II, представляют собой макроциклические пептиды, вырабатываемые грибами Amanita phalloides, и обладают высокой термостойкостью, устойчивостью к ферментативному и кислотному разложению. Фермент РНК-полимераза II является критичным компонентом основных путей клеточного метаболизма, что снижает вероятность развития механизмов резистентности [38]. В качестве цитотоксической молекулы терапевтических конъюгатов на основе антител α-амантин показал эффективность в моделях опухолей, экспрессирующих EpCAM, ПСМА и HER2 [21, 38].
Ингибиторы ДНК-топоизомеразы I
Топоизомеразы I регулируют топологическое состояние клеточной ДНК путем разрыва и последующего за релаксацией суперспиралей лигирования цепей ДНК [40, 41]. Ингибиторы ДНК-топоизомеразы I оказывают цитотоксическое действие на реплицирующиеся клетки, предотвращая повторное лигирование ДНК и приводя к повреждению ДНК и последующей гибели клеток в результате апоптоза [37]. Производное камптоте-цина дерукстекан (DX-8951f) – пример ингибитора ДНК-топоизомеразы I с благоприятными характеристиками, обладающего мощной противоопухо- левой активностью широкого спектра. Цитотоксин DX-8951f не является субстратом гена MDR1 (Multi-Drug Resistance 1) и, следовательно, может быть эффективен в лечении резистентных для эм-танзина (DM1) клеток посредством механизмов, от которых зависит отток лекарственного средства транспортерами ABC (ATP-binding cassette transporters). Сацитузумаб говитекан (IMMU-132), нацеленный на TROP2, и HER2-направленный конъюгат трастузумаб дерукстекан (DS-8201) являются двумя наиболее современными терапевтическими конъюгатами на основе камптотецина и входят в число «прорывных» лекарственных препаратов для лечения HER2-положительного метастатического РМЖ и трижды негативного рака молочной железы (ТНРМЖ) [42].
Интернализация терапевтических конъюгатов и возможные подходы к ее усилению
Канонический механизм действия конъюгатов антител с цитотоксическими агентами включает связывание с целевым антигеном, интернализацию комплекса «антиген-конъюгат» внутрь опухолевой клетки, метаболизм комплекса в лизосомах и эндосомах с последующим высвобождением цитотоксического агента. Эффективная интернализация комплекса «антиген-конъюгат» имеет решающее значение для расщепления линкера лизосомальными протеолитическими ферментами или восстановительными условиями и, следовательно, для противоопухолевой активности препарата.
Скорость интернализации HER2 может регулироваться уровнем его экспрессии, посредством связывания антител с HER2 и взаимодействием с его партнерами по димеризации. Рецептор HER2 существует в состоянии динамического равновесия между эндоцитозом и рециркуляцией на поверхности клетки. Кластеризация рецепторов на поверхности клетки может приводить к быстрой интернализации рецепторов. Связывание рецептора HER2 с антителом способствует кластеризации HER2 и эндоцитозу комплекса рецептора и антитела внутрь клетки. Интересно, что биспецифические антитела способны индуцировать кластеризацию рецептора HER2 и усиливать интернализацию по сравнению с мкАТ, нацеленными на один домен [43]. Другой фактор – высокая плотность экспрессии HER2, способствует образованию гомодимеров и олигомеров HER2, а также высокой плотности кластеризации, а следовательно, повышает скорость интернализации HER2. Так, интернализация трастузумаба наиболее эффективна в областях мембранных складок опухолевых клеток, где плотность HER2 самая высокая [44].
Однако даже при высокой экспрессии HER2 комплекс «HER2-антиген» не всегда может подвергаться интернализации и высвобождению полезной нагрузки, находясь в зависимости от конформации и расположения рецептора. Так, повышенная экспрессия рецептора эпидермального фактора роста 1 типа (EGFR) способствует образованию гетеродимеров «EGFR/HER2», уменьшает количество гомодимеров «HER2/HER2» и эффективность кластеризации HER2, что снижает как эффективность интернализации, так и противоопухолевую эффективность лекарственных препаратов на основе трастузумаба. В свою очередь, фармакологическое ингибирование EGFR посредством связывания с цетуксимабом усиливает их интернализацию и противоопухолевую активность [45].
Другим примером фармакологического воздействия усиления интернализации является способность нератиниба, необратимого ингибитора пан-HER-киназы, увеличивать полиубиквитиниро-вание и последующую интернализацию рецептора HER2 посредством диссоциации белков теплового шока 90 (HSP90) в HER2-амплифицированных или мутантных опухолевых клетках [46].
Лекарственные препараты, одобренные для клинического применения
Первым конъюгатом «антитело-лекарственное средство», одобренным FDA (U.S. Food and Drug Administration) в 2013 г. для терапии распространенного HER2-положительного РМЖ, стал трастузумаб эмтанзин (T-DM1, Kadcyla®) . Трастузумаб эмтанзин представляет собой молекулу трастузумаба, конъюгированную через нерасщепляемый малеимидный линкер малеимидометилциклогексан-1-карбоксилата (mcc) с высокоактивным цитотоксином эмтанзи-ном (DM1) в соотношении (DAR) 3,5 молекулы цитотоксина на антитело.
На основании данных двух крупных клинических исследований EMILIA и TH3RESA трастузумаб эмтанзин показан к применению при метастатическом HER2-положительным РМЖ в качестве терапии второй линии [6, 47]. Согласно рандомизированному многоцентровому клиническому исследованию III фазы EMILIA (NCT00829166), у пациентов с HER2-положительным РМЖ, прогрессирующим после терапии трастузумабом и таксаном, эффективность терапии трастузумабом эмтанзином выше по сравнению c терапией ка-пецитабином и лапатинибом (медиана ОВ – 29,9 мес [95 % ДИ 26,3–34,1] vs 25,9 мес [95 % ДИ 22,7–28,3], коэффициент риска – 0,75 [95 % ДИ 0,64–0,88]) [47]. Рандомизированное открытое исследование III фазы TH3RESA (NCT01419197), включавшее в себя пациентов, получавших ранее две или более схемы лечения, показало, что терапия трастузумабом эмтанзином приводила к значительному улучшению общей выживаемости по сравнению с лечением по выбору врача (медиана ОВ – 22,7 мес [95 % ДИ 19,4–27,5] vs 15,8 мес [13,5–18,7]; коэффициент риска – 0,68 [95 % ДИ 0,54–0,85] [6]. Данные рандомизированного многоцентрового открытого исследования III фазы KATHERINE (NCT01772472) указали на возможность расширения спектра применения трастузумаба эмтанзина [7]. Cреди пациенток с HER2-положительным РМЖ, у которых после завершения неоадъювантной терапии сохранялась опухоль, риск прогрессирования заболевания или смерти при применении трастузумаба эмтанзина в адъювантном режиме был на 50 % ниже, чем при монотерапии трастузумабом [7]. Однако при рассмотрении трастузумаба эмтанзина в качестве основной неоадъювантной терапии рандомизированное многоцентровое открытое исследование III фазы KRISTINE (NCT02131064) выявило, что трастузумаб эмтанзин в сочетании с пертузума-бом уступает традиционной неоадъювантной системной химиотерапии в комбинации с двойной HER2-блокадой трастузумабом и пертузумабом. Полный ответ на лечение был достигнут у 44,4 % больных в группе трастузумаба эмтанзина в сочетании с пертузумабом по сравнению с 55,7 % в группе доцетаксела, карбоплатина в сочетании с трастузумабом и пертузумабом (95 % ДИ от -20,5 до -2,0 p=0,016). При этом в группе трастузумаба эмтанзина в сочетании с пертузумабом реже наблюдались нежелательные явления III–IV степени (13 vs 64 %) или серьезные нежелательные явления (5 vs 29 %), чем в группе контроля [8].
Трастузумаб дерукстекан
(T-DXd, Enhertu®, ранее DS-8201a)
Новым многообещающим HER2-направленным терапевтическим препаратом, разработанным для улучшения характеристик конъюгатов «антителолекарственное средство» и одобренным FDA, является трастузумаб дерукстекан (T-DXd, Enhertu®, ранее DS-8201a). Трастузумаб дерукстекан представляет собой молекулу трастузумаба, конъюгированную с дерукстеканом (DXd) – ингибитором ДНК-топоизомеразы I – через малеимидный тетрапептидный линкер (Gly-Gly Phe-Gly), избирательно расщепляемый катепсинами, активность которых повышена в раковых клетках и в опухолевом микроокружении. После высвобождения внутри клетки цитотоксин дерукстекан связывается с расщепляемыми топоизомеразой I ДНК-комплексами и стабилизирует их, что приводит к индукции двухцепочечных разрывов ДНК и апоптозу клетки [48]. Трастузумаб дерукстекан обладает соотношением цитотоксина к антителу (DAR), равным 8, что более чем в 2 раза превышает DAR трастузумаба эм-танзина и обеспечивает мощный цитостатический эффект. Кроме того, дерукстекан обладает высокой проницаемостью мембраны, что позволяет оказывать цитотоксическое воздействие посредством «эффекта свидетеля» на опухолевые клетки в непосредственной близости от клеток-мишеней, независимо от уровня их экспрессии HER2. Данные особенности способствуют противоопухолевому эффекту препарата как при опухолях с высоким уровнем экспрессии HER2, так и в новообразованиях с низкой экспрессией [49].
Дополнительно обнаружено усиление противоопухолевого иммунитета при применении трастузумаба дерукстекана. Исследование с использованием иммунокомпетентной мышиной модели с опухолью человека, экспрессирующей HER2, на клеточной линии CT26.WT показало повышенную экспрессию CD86 на дендритных клетках, повышенную экспрессию PD-L1 и главного комплекса гистосовместимости (MHC) класса I в опухолевых клетках с последующим отторжением опухолевых клеток адаптивными иммунными клетками [50, 51].
По результатам исследования фазы II DESTINY-Breast01 (NCT03248492), трастузумаб дерукстекан одобрен в FDA в 2020 г. в рамках ускоренного одобрения для лечения больных неоперабельным или метастатическим HER2-положительным РМЖ, получивших две или более линии анти-HER2-терапии [52, 53]. В 2022 г. получены дополнительные результаты, доказывающие эффективность трастузумаба дерукстекана: частота объективного ответа (ЧОО) составила 62,0 % [95 % ДИ 54,5–69,0 %] у пациенток, получавших 5,4 мг/кг трастузумаба дерукстекана каждые 3 нед (n=184), медиана общей выживаемости (OВ) – 29,1 мес (95 % ДИ 24,6–36,1 мес), медиана выживаемости без прогрессирования (ВБП) – 19,4 мес [95 % ДИ 14,1–25,0 мес) [10].
В многоцентровом открытом рандомизированном исследовании III фазы с активным контролем DESTINY-Breast03 проведена оценка эффективности и безопасности трастузумаба дерукстекана по сравнению с трастузумабом эм-танзином. В исследование включены пациентки с HER2-положительным неоперабельным или метастатическим РМЖ, у которых наблюдалось прогрессирование в процессе лечения трастузумабом и таксанами или после проведенной терапии. Монотерапия трастузумабом дерукстеканом показала практически в 4 раза большую эффективность по сравнению с монотерапией трастузумабом эмтанзином на момент окончания сбора данных. При этом медиана ВБП в группе трастузумаба дерукстекана составила 29,0 мес (95 % ДИ 23,7– 40,0 мес) против 7,2 мес (95 % ДИ 6,8–8,3 мес) в группе трастузумаба эмтанзина [КР 0,30; 95 % ДИ 0,24–0,38]. Через 36 мес ВБП составила 45,7 % (95 % ДИ, 38,9–52,2 %) против 12,4 % (95 % ДИ 8,1–17,7 %). Наблюдалась тенденция к увеличению ОВ: медиана ОВ составила 52,6 мес (95 % ДИ 48,7 мес – нд) при применении трастузумаба деруксте-кана против 42,7 мес (95 % ДИ 35,4 мес – нд) при терапии трастузумабом эмтанзином, риск смерти был снижен на 27 % [КР 0,73; 95 % ДИ 0,56–0,94]; ЧОО составила 78,9 % (95 % ДИ 73,5–83,7 %) для трастузумаба дерукстекана по сравнению с 36,9 %
(95 % ДИ 31,0–43,0 %) для трастузумаба эмтанзина. Прогрессирование наблюдалось у 1,1 % пациенток, леченных трастузумабом дерукстеканом, и у 17,5 % больных из группы трастузумаба эмтанзина [54]. Количество нежелательных явлений ≥III степени было схожим – у 56 и 52 % больных соответственно. Характерные для трастузумаба дерукстекана лекарственно-ассоциированное интерстициальное заболевание легких или пневмонит установлены у 15 % пациенток, принимавших трастузумаб де-рукстекан, и у 3 % пациенток, получавших трастузумаб эмтанзин, при этом ни в одной из групп не было нежелательных реакций IV–V степени [11]. Исследование подтвердило удовлетворительный профиль безопасности трастузумаба дерукстекана, отсутствие кумулятивной токсичности при его применении и показало клиническое преимущество этого препарата перед трастузумабом эмтанзином, включенным в стандарты HER2-направленной терапии.
Инновационные лекарственные средства
Трастузумаб дуокармазин (SYD985) представляет собой молекулу трастузумаба, нековалентно связанную с цитотоксином дуокармицином посредством расщепляемого линкера (vc-seco-DUBA). Цитотоксический агент дуокармицин (DUBA), связываясь с малой бороздкой ДНК, необратимо алкилирует ДНК как в делящихся, так и в неде-лящихся клетках. Повреждение ДНК приводит к митохондриальному стрессу, нарушению транскрипции ДНК и апоптозу. При этом, благодаря мембранопроницаемой природе цитотоксина в комплексе с линкером, расщепляемым цистеиновыми протеазами, такими как катепсин B, присутствующими в эндосомах и лизосомах опухолевых клеток, последующее высвобождение дуокармици-на путем диффузии способствует «эффекту свидетеля», обеспечивающего активность препарата и в опухолевых клетках с низкой экспрессией HER2.
Эффективность и безопасность трастузумаба дуокармазина оценена в рандомизированном многоцентровом исследовании 3 фазы TULIP III (NCT03262935) в сравнении с препаратами, назначенными по выбору лечащего врача у пациенток с HER2-положительным местнораспространенным или метастатическим РМЖ, получивших две или более линии терапии или терапию трастузумабом эмтанзином. Медиана ВБП при терапии трастузумаба дуокармазина значительно отличалась и составила 7,0 мес [95 % ДИ 5,4–7,2] по сравнению с 4,9 мес [95 % ДИ 4,0–5,5] при лечении по выбору врача. Наиболее частыми побочными эффектами трастузумаба дуокармазина явились офтальмологическая токсичность (конъюнктивит –38,2 %, кератит – 38,2 %) и интерстициальные заболевания легких/пневмонит – 7,6 % [13].
ARX788 (Ambrx Inc.). Конъюгат нового поколения, разработанный для обеспечения гомогенности и химической стабильности, ARX788, представляет собой HER2-направленное моноклональное антитело, ковалентно связанное с ингибитором тубулина амберстатином (AS269, является запатентованной версией монометилауристатина F (MMAF)) с соотношением лекарственного средства к антителу (DAR) 1,9. Традиционное конъюгирование антитела и цитотоксина предполагает использование нативных, экспонированных на поверхности антитела, лизинов или цистеинов. Неспецифическая конъюгация приводит к образованию смеси с переменным соотношением лекарственного средства к антителу (DAR) и неопределенными сайтами конъюгации, которые часто нестабильны в пуле крови. От других разработанных терапевтических конъюгатов ARX788 отличает использование технологии сайт-специфической оксимной конъюгации посредством включения в антитело неприродной аминокислоты пара-ацетилфенилаланина (pAF), которая лишает препарат описанных выше недостатков [55]. Другой отличительной чертой является наличие нерасщепляемого высокогидрофобного линкера с ограниченной клеточной проницаемостью. Стабильный линкер снижает системную токсичность разрабатываемого препарата, но лишает эффекта «уничтожения свидетеля», который используется в таких HER2-нацеленных конъюгатах, как трастузумаб дерукстекан и трастузумаб дуокармазин, для цитотоксического воздействия на опухолевые клетки с низким уровнем HER2. В то же время доклинические исследования показали высокую активность ARX788 в опухолях с различной экспрессией HER2, а также T-DM1-резистентных новообразованиях [56]. Клиническое исследование I фазы подтвердило эффективность и низкую системную токсичность ARX788. При рекомендуемой дозе для II фазы 1,5 мг/кг частота объективного ответа составила 65,5 % [95 % ДИ], а медиана выживаемости без прогрессирования – 17,02 мес (95 % ДИ, 10,09 – не достигнуто) [16]. На данный момент продолжается рандомизированное исследование II фазы у HER2-положительных пациенток с метастатическим раком молочной железы ACE-Breast-03 (NCT04829604).
Занидатамаб зоводотин (ZW49). В новом исследуемом конъюгате занидатамабе зоводадотине (ZW49) молекулой-носителем является биспецифическое моноклональное антитело занидатамаб (ZW25), нацеленное на II и IV субдомены HER2 (субдомены, на которые нацелены пертузумаб и трастузумаб соответственно). Занидатамаб способствует кластеризации рецепторов, при которой каждый рецептор HER2 может быть мишенью для двух молекул антител занидатамаба [57]. Занида-тамаб нековалентно связан через расщепляемый линкер с ингибитором сборки тубулиновых микротрубочек, монометилауристатином E (MMAE). Доклинические данные продемонстрировали ускоренную интернализацию занидатамаба зо- водотина по сравнению с конъюгатами на основе моноспецифического трастузумаба, а также противоопухолевый эффект на клеточных моделях с высокой и низкой экспрессией HER2, моделях с метастазами в головной мозг [58]. Безопасность и переносимость ZW49 оцениваются в I фазе клинического исследования при метастатическом РМЖ, экспрессирующем HER2 (NCT03821233) [17].
MRG002 – новый HER2-направленный конъюгат, состоящий из модифицированного сахаром трастузумаба и разрушающего микротрубочки агента монометилауристатина E (ММАЕ), соединенных через расщепляемый валин-цитруллиновый (vc) пептидный линкер. В доклинических исследованиях доказана эффективность MRG002 при HER2-положительном РМЖ, а также при РМЖ с низкой экспрессией HER2. Конъюгат продемонстрировал схожую с трастузумабом аффинность связывания, но значительно более низкую активность антителозависимой клеточной цитотоксичности (ADCC). В дополнение к мощной цитотоксичности in vitro , применение MRG002 вызывало регрессию опухоли в моделях ксенотрансплантатов in vivo с высокой и средней/низкой экспрессией HER2. Кроме того, установлена повышенная противоопухолевая активность при использовании MRG002 в сочетании с антителом против рецептора PD-1 (Programmed cell death-1, CD279) [59]. На данный момент в Китае проходят клинические исследования II фазы MRG002 в качестве терапии пациенток с HER2-положительным неоперабельным местнораспространенным или метастатическим РМЖ, получавших ранее терапию трастузумабом и ингибитором тирозинкиназы HER2, у пациенток с местнораспространенным или метастатическим РМЖ с низким уровнем HER2, а также у больных HER2-положительным неоперабельным местнораспространенным или метастатическим раком уротелия [18].
Диситамаб ведотин (RC48, Aidixi ® ) так же, как и MRG002, представляет собой мАТ, связанное через расщепляемый валин-цитруллиновый линкер с цитотоксическим агентом MMAE. Отличительной чертой является использование в качестве носителя мАТ гертузумаба, воздействующего на различные эпитопы рецептора HER2.
FS-1502 представляет собой HER2-напра-ленный конъюгат на основе мкАТ, аналога трастузумаба, и цитотоксического агента MMAF, ингибирующего полимеризацию тубулина, соединенных через расщепляемый β-глюкуронидный линкер с соотношением DAR=2. Для данного терапевтического конъюгата разработан новый запатентованный метод сайт-специфичной конъюгации и высокостабильный линкер. Продемонстрировано, что сайт-специфичная конъюгация с низким значением DAR=2 превосходит другие терапевтические конъюгаты с более высоким DAR благодаря меньшей агрегации, медленному системному клиренсу и меньшей токсичности. FS-1502 продемонстрировал эффективную остановку клеточного цикла и повышенную HER2-направленную цитотоксичность по сравнению с трастузумабом эмтанзином в доклинических исследованиях in vitro . На данный момент продолжается многоцентровое одногрупповое открытое клиническое исследование фазы I с фазой эскалации дозы для оценки безопасности и переносимости FS-1502 у пациентов с HER2-выраженными злокачественными солидными опухолями на поздних стадиях (фаза 1a) и с расширенной когортой (фаза 1b) для оценки эффективности FS-1502 у пациенток с метастатическим HER2-положительным РМЖ.
По предварительным опубликованным данным, ЧОО – 53,7 % (95 % ДИ, 41,1–66,0 %), включая подтвержденную ЧОО, равную 37,5 %. FS-1502 также показал противоопухолевую активность при РМЖ с низкой экспрессией HER2, ЧОО составила 26,1 %. Наиболее частыми НЯ ≥III степени были гипокалиемия (15,3 %) и снижение количества тромбоцитов (8,0 %), у 3,3 % пациентов наблюдался пневмонит; у 2,7 % – обратимые глазные явления III степени. В целом, FS-1502 характеризовался хорошей переносимостью и продемонстрировал многообещающую противоопухолевую эффективность при HER2-положительном РМЖ [19].
BL-M07D1 – конъюгат HER2-направленного мкАТ и нового ингибитора топоизомеразы I (Ed-04), соединенных линкером, расщепляемым катепсином B. Соотношение DAR составляет 8. В доклинических исследованиях in vivo BL-M07D1 продемонстрировал более высокую противоопухолевую активность в различных моделях HER2-экспрессирующих опухолей по сравнению с трастузумабом эмтанзином и трастузумабом дерукстеканом. При этом BL-M07D1, продемонстрировав выраженный «эффект свидетеля», оказался более эффективным в моделях in vivo с низкой экспрессией HER2 и смешанных моделях ксенотрансплантатов HER2-положительных/ отрицательных по сравнению с трастузумабом дерукстеканом. На данный момент проводится фаза I клинического исследования для изучения
