Structural Protein Function Prediction - A Comprehensive Review

Author: Huda A. Maghawry, Mostafa G. M. Mostafa, Mohamed H. Abdul-Aziz, Tarek F. Gharib
Journal: International Journal of Modern Education and Computer Science (IJMECS) @ijmecs
Article in issue: 10 vol.7, 2015.
Free access
The large amounts of available protein structures emerges the need for computational methods for protein function prediction. Predicting protein function is mainly based on finding similarities between proteins with unknown function with already annotated proteins. This may be achieved using different protein characteristics: sequences, interactions, localization, structure and or psychochemical. A lot of review papers mainly focus on sequence and psychochemical features-based methods. This is because sequence and psychochemical data are easy to deal with and to interpret the results, and much available compared to protein structures. However, structure-based computational methods provide additional accuracy and reliability of protein function prediction. Therefore, unlike many review papers, this paper presents an up-to-date review on the structure-based protein function prediction. The aim was to provide a recent and comprehensive review of protein structure related topics: function aspects, structural classification, databases, tools and methods.
Protein function prediction, protein structure, structure alignment, comparison, distance matrix, binding sites, classification
Short address: https://sciup.org/15014804
IDR: 15014804
Text of the scientific article Structural Protein Function Prediction - A Comprehensive Review
Published Online October 2015 in MECS DOI: 10.5815/ijmecs.2015.10.07
Applying computer science techniques to biology – bioinformatics - caused a pounce in automated prediction of protein function. Proteins are the basis of cellular life. Their importance emerged from that they significantly affect the structural and functional characteristics of living cells. Proteins may have different representations varied from its sequence to structure. Protein structures are highly complex and have a high range of variability. Identification of the protein structures and functions are most important for the treatment of diseases and drug industry. There are millions of available protein structures from many of high-throughput genome projects [1, 2]. A few numbers of these proteins have been experimentally annotated [3]. This experimental function annotation of newly discovered proteins is achieved with very small throughput and high cost [3] but using computational methods can infer the protein function with very high throughput and lower cost. Therefore, efficient and accurate computational methods for protein function prediction are highly required. Using computer science methods in order to predict protein functions is known as computational function prediction, computational function annotation or computational proteomics [4]. Most of current review papers give a little insight on the effectiveness of using protein structures in function prediction. Therefore, the aim of this review is to focus on structure-based protein function prediction. First, historical background about how the protein structures characterized, are mentioned with the latest statistics related to the depositions and the growth of the structures. Then, both leading and recent structural classifications of protein structures are reviewed. Structure comparisons and finding similarities [5] for function prediction is classified according to the level of comparison into global structure comparison-based approach and local structure comparison-based approach. Global structure comparison-based approach considers the whole protein and uses protein geometry and/or secondary structure elements (SSEs) features. Local structure comparisonbased approach considers substructures and used for finding conserved regions or predicting sites that are significant in deriving protein function. Finally for both approaches, popular besides recent databases, online servers, tools and methodologies are reviewed in terms of accuracy and limitations.
Most of computational protein function prediction research methodology begins with one of two directions: studying how to find an effective protein representation method or proposing an accurate algorithmic prediction method. Regarding proposing a protein representation, it mostly starts with testing it on a benchmark and rarely on newly-built dataset related to a certain function prediction aspect. Testing may be accomplished using certain classifier such as the frequently used classifier SVM as it is easier to implement and proved to be powerful classifiers for protein function prediction. However, its disadvantages are that they give little information about the patterns learned and couldn’t be generalized to other datasets than used in their training, or different classifiers. Besides, representation proposal may be extended to include combinations of the proposed representation and other published representations that proved their efficiency in prediction. If the proposed representation achieved an improvement, tests may be extended to other datasets or even other aspects of protein function prediction. Regarding the direction of proposing a prediction algorithmic method, it may be a combination of known classifiers as the performance of ensemble machine learning approaches is much better than the performance of the individual learning algorithm, or proposing new one. The method usually utilizes available protein representations. If the proposed method proved its efficiency, tests may be extended to different datasets or other aspects of protein function prediction to investigate the method significance. Fig. 1 summarizes protein function prediction framework.
|
Proteins Dataset |
Proteins Representation |
Prediction Method |
Evaluation |
||||
|
Raw Form or Features |
Classifier or Ensemble |
||||||
|
Benchmark or Extraction and/or of Classifiers ж Features Selection New Dataset |
|||||||
Fig.1. The Protein Function Prediction Framework.
The huge amount of biological data has to be stored, analyzed, and retrieved. Protein databases are categorized as primary or structural [6]. Primary protein databases contain protein sequences. Example of these databases is SWISS-PROT [7]. SWISS-PROT annotates the sequences as well as describing the protein functions. Structural databases contain molecular structures. The protein data bank PDB [8] is the main database for three dimensional structures of molecules specified by X-ray crystallography and NMR (nuclear magnetic resonance). The PDB entries contain the atomic coordinates, and other structural atomic and secondary structure elements related attributes. The first bio-macromolecular NMR structure was archived on 1989. Fig. 2 shows the yearly growth in PDB structures. The world wide PDB published statistics showing the PDB structures that are deposited by year (Fig. 2) and current PDB holdings by year (Fig. 4).
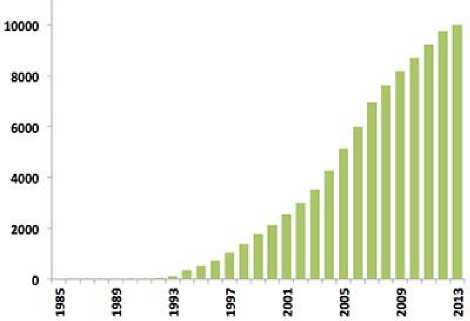
Fig.2. Yearly Growth of NMR-Derived Structures in the PDB []
-
II. Protein Structural Classifications
There are two major leading structural classifications of proteins: SCOP [9] and CATH [10]. The concept of protein structural classes was reported by Levitt and Chothia [11], which grouped proteins based on their predominant secondary structural element. Structural Classification of Proteins (SCOP) database structurally classifies proteins into four levels of hierarchical classification: structural class, fold, super family and family based on the structural and evolutionary relationships. It is based on the manual classification therefore, its classification is considered accurate. However, an extended version of SCOP is now available [12] to automatically classify structures with maintaining the same accuracy level.
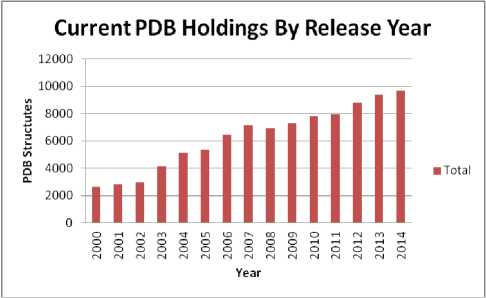
Fig.3. Statistics for PDB Structures Depositions Yearly (Last updated at 30 Dec 2014).
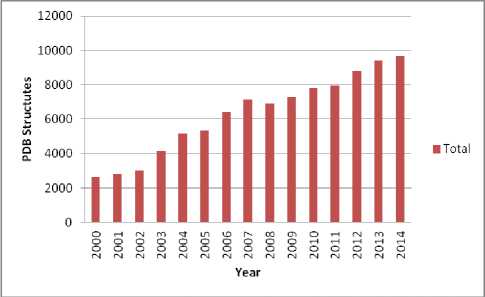
Fig.4. Current PDB Holdings by Release Year (Last updated at 30 Dec 2014).
The CATH domain structure database: The four main levels of CATH classification are C for protein class, A for architecture, T for topology and H for homologous superfamily. The classification is based on the secondary structure, composition, orientation and/or connectivity. However, a recent improvement to family classification is applied in CATH [13].
Recently, a structural classification of loops in proteins (ArchDB) [14] and an evolutionary classification of protein domains (ECOD) [15] are presented. Unlike structural classification that assumes proteins to be evolutionarily related according to homology, ECOD uniquely focuses on remote homology that is difficult to detect. ECOD classified more than 107,000 PDB structures and is weekly updated to include new released PDB structures.
ArchDB is a structural classification of loops extracted from known protein structures. It includes 10 different loop types based on the geometry and the conformation of the loop.
-
III. Structure-Based Function Prediction Methods
Proteins functions are related to their structural role or enzymatic role [16]. The structural role is related to forming the cell shape. The enzymatic role is related to help in accomplishing chemical reactions, signal movement in and out of the cell and transportation of different kind of molecules like antibodies, structural binding elements, and movement-related motor elements. The computational approaches predict the function of a given protein by searching for similarities between its structure and other functionally annotated proteins using different techniques. Hence, the functional annotation of the most similar protein is transferred to the query protein. Aiming at predicting functions of proteins, analyzing protein structures that belong to a certain functional class, help in deriving specific structural features that are highly conserved within this functional class. These structural features can then be studied to identify classes of a new protein structure as well as its function. Annotating proteins is the primary goal of function prediction computational methods. Therefore, the available biological knowledge should be in a form that is applicable with computational processing. Different annotations are used to describe proteins function: enzyme commission (EC) numbers [17] and Gene ontology consortium (GO) [18]. EC numbers [17] use four digits for enzyme classification based on the reactions they catalyze. GO [18] describes protein functions with respect to its molecular function, biological process, and cellular component. In some cases, prediction of other protein properties may help in inferring and understanding its function like subcellular localization; which means where a protein resides in a cell [19] and fold recognition; as proteins having similar structural folds may share the same functions.
With the availability of protein structures having experimented annotations, predicting protein function by finding structural similarity between them and the protein structures that functionally un-annotated becomes possible even if their sequences are not similar [20]. Structure-based protein function prediction methods are based on identifying similarity between a protein with no knowledge about its function and one or set of proteins with known function using structural features. These computational methods are based on statistical, data-mining and/or machine learning techniques [21-23]. In the following subsections, we will review recent tools and computational methods for structure comparison and function prediction. The essential step related to any of proteins function prediction methods is how the protein should be effectively represented. Effective representations improve the prediction accuracy and minimize the information loss. Structure-based function prediction approaches can be categorized according to the level of protein structure comparison into global structure comparison-based approach or local structure comparison-based approach. Global structure comparison based approaches are mainly based on proteins atoms coordinate or secondary structure elements. While, local structure comparison based approaches are mainly based on the surface shape. For more reviews of function prediction approaches see [24-29].
There are online servers available for predicting protein function from structure. ProFunc [30] is a protein biochemical function prediction server
. It applies various methods, including fold matching, residue conservation, and 3D functional templates. Then, a list of the probable functions in terms of GO annotations is provided. Recently, I-TASSER [31] (Iterative Threading ASSEmbly Refinement) was ranked the best for function prediction in CASP9 (critical assessment of protein structure prediction) [32] experiment. The server, which is available at is aiming to implement the state-of-the-art structure and function prediction algorithms in order to provide the most accurate results.
-
A. Global-Structure Comparison-based Approach
This approach is based on analyzing the whole protein structure and find relations to other protein structures with known function [33]. Then, this relation is used to predict the functions of functionally un-annotated proteins. Finding similarities between pairwise or multiple protein structures requires structural comparison which may require alignment technique [34, 35]. This is will not be achieved without developing an efficient proteins structure representation. Therefore, global-structure comparison-based approaches are divided into two categories based on the protein structural representation. They may be based on proteins geometric [36-44] or secondary structure elements (SSEs) [45-51].
Regarding methods that based on protein geometry, the much known structural alignment methods are DALI [44] and CE [43]. In DALI or Distance-matrix ALIgnment each protein is represented as a 2D matrix storing intra molecular distance. CE or Combinatorial Extension method represents a protein as a set of Cα distances between eight consecutive residues. Different methods [40-41] were proposed which perform faster with high accuracy levels than DALI and CE. They apply dynamic programming like ALYDYN [41] and TM-align [42]. CLICK [40] is a tool which capable of using other structural features besides protein coordinates to align structures by matching cliques of residues.
Regarding methods based on SSEs, the much known structural alignment methods are SSAP [50] (or Sequential Structure Alignment Program) and VAST [51] (Vector Alignment Search Tool). Recently, TS-AMIR [46] or Topology String Alignment Method for Intensive Rapid comparison of protein structures which is proposed in order to reduce the complexity of the structure comparison process. They represent the protein based on the secondary structure elements (SSEs) of its backbone structure. DecosnSTRUCT [47] is a database search, and pairwise alignment method that uses a reduced protein representation based on the direction, type and sequential ordering of SSEs as features for comparisons [48]. Table 1 presents both popular and recent tools and their URLs.
Recently, advanced tools were proposed for macromolecular complexes. Examples of geometry-based tools are TopMatch [39] and TopSearch [36] and contact area difference score web server (CAD-score) [37]. Example of SSEs-based tools is VAST+ [45]. Several tools are reviewed in [52].
Table 1. List of Recent and Popular Structural Comparison and Alignment Tools.
Tool URL CAD-score [37] MICAN [38] TopMatch [39] CLICK [40] DALI server [53] ALADYN [41] CE (RCSB PDB) [54] TM-ALIGN [42] SSAP [50] VAST+ [45] deconSTRUCT [47]
The widely used protein representation is based on atom-coordinates. Such representation mainly uses Cα atoms which known as protein backbone, to represent the whole protein structure rather than using all atoms to lower the computational complexity. Each protein can be transformed into a matrix of distances between all its Cα atoms which is known as distance matrix. Since it has been used with DALI, different variations of distance matrix are still being proposed. Examples of these variations are: Contact maps [55, 56] and Cutoff Scanning Matrix (CSM) [57]. Contact map is a matrix contains Boolean values representing pairwise inter residue contact for a protein structure. The matrix values are determined according to a threshold distance. Bhavani et al. [58] used contact maps to predict protein folds. Based on triangle subdivision method and using decision tree for binary classification, they correctly predicted EF-hand-like and cytochrome fold with accuracy of 96% and 79%, respectively. CSM [57] is a distance based protein structure representation which generates feature vector that represents distance patterns between protein residues using different threshold values. Adding other features to CSM resulted in new protein representation, PSM-C [59]. PSM-C builds the protein feature vector by including inter-residue angle and distance patterns. PSM-C representation achieved accuracy levels higher than CSM (10% in average) in predicting superfamily and family and in discriminating enzyme proteins using Random Forest. Table 2 summarizes the results obtained.
Some limitations of distance matrix are its sensitivity to the parameters and its high dimensionality. Wavelets were proposed to reduce the distance matrix dimensionality [60] and extract features [61]. Marsolo and Ramamohanarao [60] reduce the dimensionality of protein structure by generating a feature vector of approximation coefficients by applying 2- dimensional wavelet decomposition of the distance matrix which allows fast retrieval of similar structures. An average accuracy of 87% was achieved at SCOP superfamily level using a k-d tree and a 10-nearest-neighbor on a dataset consists of 33,000 proteins. Recently, Mirceva et al. [61] performed different wavelet transforms on the protein distance matrix and found that Daubechies2 wavelet gives the highest retrieval accuracy of 91.3%.
Table 2. Summary of Computational Prediction Methods That are Based on Distance Matrix.
|
Method |
Ref. |
Application |
Performance |
|
Decision Tree |
[58] |
EF-hand-like Prediction |
96%* |
|
Decision Tree |
[58] |
Cytochrome Prediction |
79%* |
|
Random Forest |
[59] |
Enzyme Discrimination |
79.25%* |
|
Random Forest |
[59] |
Superfamily Prediction |
98%* |
|
KNN |
[57] |
Superfamily Prediction |
94.2%** |
|
Random Forest |
[59] |
Family Prediction |
91%* |
* Accuracy, ** Precision
-
B. Local-Structure Comparison-Based Approach
Local-structure comparison-based approach differs from the previous approach that only substructures are analyzed. This includes finding motifs or conserved regions in proteins sharing the same function which is significant in function analysis. It also includes finding specific regions related to functions called protein functional sites such as binding sites [62]. Examples of the recent online resources for substructures databases are BioLiP database [63] and ProBiS [64]. PDB doesn’t present all biologically relevant ligands. Therefore, BioLiP database is developed for biologically relevant ligand–protein interactions. It includes manual verification phase. The latest version of BioLiP (Feb 27, 2015) includes 308,776 entries. Each entry is annotated using ligand-binding, residues, EC numbers, GO terms. ProBiS database contains nearly 420 million searchable pre-calculated pairwise alignments. It includes over 37600 locally structurally aligned non-redundant PDB structures to all other proteins. The sever is also used to identify functionally significant binding-site residues, detect weak similarities in proteins with non-similar folds, deriving functional annotation, finding similar binding- sites in proteins of different families. Table 3 provides databases and their URLs.
Table 3. Protein Substructure Databases.
Resource Short Description URL BioLiP [63] Ligand-protein binding database ProBiS [64] Repository for structurally similar protein binding sites CASTp [65] Protein pockets and cavities server Catalytic Site Atlas [66] Database of enzyme active sites and catalytic residues in enzymes on-srv/databases/CSA/ eF-site [67] Binding sites database
Motif finding can be achieved by different methodologies. These include representing motifs based on SSEs features [49], representing proteins as graphs and motifs as sub-graphs [68], transforming the structure coordinates into alphabet sequences [69], representing motif using distance matrix [70] or based on structure spatial arrangement [71].
ProSMoS server [49] or protein structure motif search used SSE types, connectivity, coordinates, interactions type to search for a motif. Jia et al. [68] applied graph algorithm, AProximate Graph Mining (APGM), to find repeated sub-graphs and hence conserved substructures and achieved an accuracy of 78%. Ku and Hu [69] used sequence-based tool to find motifs after transforming protein structure coordinates into alphabet sequences. A Distance matrix is also used by [70] to represent the spatial structure of the domain which is the functional unit of the whole protein, and perform fast domain classification with an accuracy of 90%. Based on spatial arrangements, Rahimi et al. [71] searched for the representative motif for each EC number.
Regarding sites prediction, these substructures may have a similar surface shape. Therefore, identifying unknown functional sites achieved by searching for similar structures related to functional sites in proteins with known function [72, 73]. Binding sites are where proteins function by binding to another protein. Nisius et al. [74] provide a review of methods for binding site prediction in terms of accuracy and limitations. There are available web servers for site prediction. The popular one is eF-seek [67]. eF-seek search finds the similar ligand binding sites. Recently, 3DligandSite [75], SPRITE [76] and mentioned earlier ProBiS web servers become available for binding-site prediction. 3DligandSite makes use of protein-structure prediction to model unsolved proteins. It searches for similar structures, then superimpose ligands bound to the model and used to predict the binding site. 3DligandSite provides conservation details like the predicted binding-site residues list with details of the number of ligands that they contact. SPRITE (Search for protein sites) aims to infer the functions by searching for matches in the 3D patterns of amino acid side chains based on graph theory. ProBiS offers comparison of a binding site, pairwise alignment and superimposition of PDB structures.
Table 4. List of Popular and Recent Structure-Based Binding Sites Prediction Tools.
Tool Ref. URL eF-seek [67] 3DLigandSt ie [75] SPRITE [76] ProBiS [64]
Several structure based prediction methods were proposed to predict binding sites in proteins. They mainly based on representing proteins using shape descriptor as in [77] that identified accurately 85% of known binding sites using alpha carbon atom of each residue. 3D Zernike descriptor introduced by [78], represents a protein structure as a series expansion of 3D functions. The advantages of using 3D Zernike that it allows fast protein structures retrieval, it is rotation invariant as no alignment is needed for protein structure comparison and it can be adjusted to the different resolutions of protein structures description [79]. For more readings in 3D Zernike descriptor, see [80], for more moments-based descriptors see [81]. Other methods are based on structural features as in Zhao et al. [82] that made use of knowledge-based energy function and atom-type-dependent features and correctly predicted 98% of DNA binding proteins. Spin Images which also used for three-dimensional object recognition in computer vision area are proposed by [83] to represent protein surfaces as a set of two dimensional images. Moment-based descriptors have lower time complexity than graph methods and spin images. Results are summarized in Table 5.
Table 5. Results Summary of Local-Structure Comparison-Based.
|
Methodology |
Ref. |
Application |
Accuracy |
|
AProximate Graph Mining (APGM) |
[68] |
Finding conserved structures |
78% |
|
Shape Descriptor |
[77] |
Binding site prediction |
85% |
|
Structural Features |
[82] |
DNA Binding site prediction |
98% |
|
Spatial Structure |
[70] |
Domain Classification |
90% |
-
IV. Discussion
There is a continuous and rapid growth in protein structures. New NMR Protein structures that are deposited the last year were more than combined NMR depositions in the first 10 years since 1989. Protein structures are usually classified to derive their function annotation. The two recent structural classification of
protein considers two important factors missed in the popular leading classifications: remote homology between proteins and loop classification. Protein representations, its computational cost and dimensionality directly, affect the efficiency and accuracy of the protein function prediction method. Regarding function prediction based on global structure comparison, most of available online servers are aiming to provide fast and the most accurate results for structure comparison and function prediction. CLICK [40], ALYDYN [41] and TM-align [42] are example of geometry-based methods in literature that are faster with high accuracy levels than popular CE and DALI. CLICK is statistically better than DALI in terms of structure overlap. ALYDYN can align in less than 1 minute proteins with up to 250 amino acids. TM-align is faster than DALI with 20 times and CE with 4 times. However, SSEs-based methods [46, 47] are faster than geometry methods because they depend on reduced and simpler representation of proteins. TS-AMIR [46] runs hundreds of times faster than geometry based methods CE and TM-Align while maintaining the same accuracy levels. Deconstruct handles a test query data in only 2 minutes, which is faster than CE and Top Match which takes 2,915 minutes and 67 minutes, respectively. Based on the distance matrix concept, decision trees achieved range of overall accuracy from 79% to 98% (Table 2). The cons of distance matrix are its sensitivity to construction parameters and its high dimensionality. However, such limitations can be solved using Wavelets to reduce dimensionality and extract features. Regarding function prediction based on local structure comparison, shape descriptors which consider overall protein surface shape features achieved higher prediction accuracy than SSEs features. Surface shape based approaches are proving to be more accurate and biologically meaningful with binding and functional sites. However, they are computationally higher than utilizing the residues coordinates. Some structure based methods utilize evolutionary information with structure information to improve the accuracy of prediction. Till now, no one exact method or one exact representation can be decided to be the best for predication. Accurate and datasetindependent methods with lower computational complexity are still needed for different aspects of protein function prediction.
-
V. Conclusion
Using computational methods for the prediction of protein functions became essential. It saves much laboratory needed effort, time and cost. They may be based on any of protein characteristics, i.e. sequences or structures. However, protein functions are highly related to their structures. Therefore, structure-based function prediction methods attract a great attention. They are highly required and challenging in the function analysis of proteins. Therefore, this review provides both leading and recent structural classifications, function prediction methods, databases and tools that are based on protein structures.
References Structural Protein Function Prediction - A Comprehensive Review
- E.W. Sayers, T. Barrett, D.A. Benson, et al. "Database resources of the national center for biotechnology information," Nucleic Acids Res, vol. 40, pp. D13–D25, 2012.
- H. Vuong, R.M. Stephens and N. Volfovsky, "AVIA: An interactive web-server for annotation, visualization and impact analysis of genomic variations," BMC Proceedings, vol. 6, pp. 37, 2012.
- D. Barrell, E. Dimmer, R. P. Huntley, D., Binns, C. O'Donovan and R. Apweiler, "The GOA database in 2009—an integrated gene ontology annotation resource," Nucleic Acids Res, vol. 37, pp. D396–D403, 2009.
- T. Hawkins and D. Kihara, "Function prediction of uncharacterized proteins," J Bioinform Comput Biol, vol. 5, pp. 1-30, 2007.
- S. Erdin, A. M. Lisewski and O. Lichtarge, "Protein function prediction: towards integration of similarity metrics," Curr Opin Struc Biol, vol. 21, pp. 180–188, 2011.
- S.C. Rastogi, P. Rastogi and N. Mendiratta, Bioinformatics Methods and Applications: Genomics Proteomics and Drug Discovery. 3rd edition. PHI Learning Pvt. Ltd. 2008.
- B. Boeckmann, A. Bairoch, R. Apweiler, et al. "The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003," Nucleic Acids Res, vol. 31, pp. 365–370, 2003.
- H. M. Berman, J. Westbrook, Z. Feng, et al. "The protein data bank," Nucleic Acids Res, vol. 28, pp. 235–242, 2000.
- A. B. Murzin, "SCOP: a structural classification of proteins database for the investigation of sequences and structures," J Mol Biol, vol. 247, pp. 536–540, 1995.
- C. A. Orengo, A. D. Michie, S. Jones, D. T. Jones, M. B. Swindells and J. M. Thornton, "CATH--a hierarchic classification of protein domain structures," Structure, vol. 5, pp. 1093–1108, 1997.
- M. Levitt and C. Chothia, "Structural patterns in globular proteins," Nature, vol. 261, pp. 552–558, 1976.
- N. K. Fox, S.E. Brenner and J.M. Chandonia, "SCOPe: structural classification of proteins—extended, integrating SCOP and ASTRAL data and classification of new structures," Nucleic Acids Res, vol. 42, D304-309, 2014.
- I. Sillitoe, A. L. Cuff, B. H. Dessailly, N. L. Dawson, N. Furnham, D. Lee, J. G. Lees, T. E. Lewis, R. A. Studer, R. Rentzsch, C. Yeats, J. M. Thornton and C. A. Orengo, "New functional families (FunFams) in CATH to improve the mapping of conserved functional sites to 3D structures," Nucleic Acids Res, vol. 41(Database issue): D490–D498, 2013).
- J. Bonet, J. Planas-Iglesias, J. Garcia-Garcia, M. A. Marín-López, N. Fernandez-Fuentes and B. Oliva, "ArchDB 2014: structural classification of loops in proteins," Nucleic Acids Res, vol. 42 (Database issue), D315-9, 2014.
- H. Cheng, R. D. Schaeffer, Y. Liao, L. N. Kinch, J. Pei, S. Shi, B. Kim and N. V. Grishin, "ECOD: an evolutionary classification of protein domains," Plos Comput Biol, vol. 10, e1003926, 2014.
- F. J. Burkowski, Structural Bioinformatics An algorithmic Approach. Chapman and Hall/CRC Mathematical & Computational Biology Series, 2009.
- D. E. Almonacid, E. R. Yera, J. B. O. Mitchell and P. C. Babbitt, "Quantitative comparison of catalytic mechanisms and overall reactions in convergently evolved enzymes: implications for classification of enzyme function," Plos Comput Biol, vol. 6, No. 3, pp. e1000700, 2010.
- M. Ashburner, C. A. Ball, J. A. Blake, et al. "Gene ontology: tool for the unification of biology, The Gene Ontology Consortium," Nat Genet, vol. 25, pp. 25–29, 2000.
- Z. P. Feng, "An overview on predicting subcellular location of a protein," In Silico Biol, vol. 2, pp. 291-303, 2002.
- D. L. Wild and M. A. S. Saqi, "Structural proteomics: inferring function from protein structure," Current Proteomics, vol. 1, No. 1, pp. 59–65, 2004.
- J. Han and M. Kamber, Data Mining: Concepts and Techniques. San Francisco, CA: Elsevier, 2008.
- P. Larranaga, B. Calvo, R. Santana, R. et al. "Machine learning in bioinformatics," Brief Bioinform, vol. 7, No. 1, pp. 86–112, 2006.
- W. Ewens and G. Grant, "Statistical methods in bioinformatics: an introduction," in Statistics for biology and health, M. Gail, K. Krickeberg, J. Samet, A. Tsiatis, and W. Wong, Eds. 2nd ed., Springer, 2005.
- A. K. Tiwari and R. Srivastava, "A survey of computational intelligence techniques in protein function prediction," International Journal of Proteomics, vol. 2014, 2014.
- A. Cuff, O. Redfern, B. Dessailly and C. Orengo, "Exploiting protein structures to predict protein functions," in Protein Function Prediction for Omics Era, D. Kihara, Ed. USA: Springer, 2011.
- O. C. Redfern, B. Dessailly and C. A. Orengo, "Exploring the structure and function paradigm," Curr Opin Struct Biol, vol. 18, pp. 394–402, 2008.
- D. Lee, O. Redfern and C. Orengo, "Predicting protein function from sequence and structure," Nat Rev Mol Cell Bio, vol. 8, pp. 995–1005, 2007.
- J. D. Watson, R. A. Laskowski and J. M. Thornton, "Predicting protein function from sequence and structural data," Curr Opin Struct Biol, vol. 15, pp. 275–284, 2005.
- G. J. Bartlett, A. E. Todd and J. M. Thornton, "Inferring protein function from structure," in Structural Bioinformatics, P. E. Bourne and H. Weissig, Eds. Hoboken, New Jersey: Wiley-Liss, 2003.
- R. A. Laskowski, J. D. Watson and J. M. Thornton, "ProFunc: a server for predicting protein function from 3D structure," Nucleic Acids Res, vol. 33, pp. W89-W93, 2005.
- J. Yang, R. Yan, A. Roy, D. Xu, J. Poisson and Y. Zhang, "The I-TASSER Suite: Protein structure and function prediction," Nat Methods, vol. 12, pp. 7-8, 2015.
- D. Xu, J. Zhang, A. Roy and Y. Zhang, "Automated protein structure modeling in CASP9 by I-TASSER pipeline combined with QUARK-based ab initio folding and FG-MD-based structure refinement," Proteins: Struct, Func, Bioinf, vol. 79, (Suppl 10), pp. 147-160, 2011.
- M. Boaretoa, M. Yamagishib, N. Catichaa, and V. Leite, "Relationship between global structural parameters and Enzyme Commission hierarchy: Implications for function prediction," Comput Biol Chem, vol. 40, pp. 15–19, 2012.
- R. Wang and S. C. Schmidler, "Bayesian multiple protein structure alignment," Research in Computational Molecular Biology. Lecture Notes in Computer Science, vol. 8394, pp. 326-339, 2014.
- D. W. Ritchie, A. W. Ghoorah, L. Mavridis and V. Venkatraman, "Fast protein structure alignment using Gaussian overlap scoring of backbone peptide fragment similarity," Bioinformatics, vol. 28, pp. 3274–3281, 2012.
- M. Wiederstein, M. Gruber, K. Frank, F. Melo and M. J. Sippl, "Structure-based characterization of multiprotein complexes," Structure, vol. 22, pp. 1063–1070, 2014.
- K. Olechnovič and Č. Venclovas, "The CAD-score web server: contact area-based comparison of structures and interfaces of proteins, nucleic acids and their complexes," Nucleic Acids Res, vol. 42 (Web Server issue), W259-W2, 2014.
- S. Minami, K. Sawada and G. Chikenji, "MICAN: a protein structure alignment algorithm that can handle Multiple-chains, Inverse alignments, Ca only models, Alternative alignments, and Non-sequential alignments," BMC Bioinformatics, vol. 14, 24, 2013.
- M. J. Sippl and M. Wiederstein, "Detection of spatial correlations in protein structures and molecular complexes," Structure, vol. 20, pp.718–728, 2012.
- M. N. Nguyen, K. P. Tan and M. S. Madhusudhan, "CLICK - Topology independent comparison of biomolecular 3D structures," Nucleic Acids Res, vol. 39, Issue suppl 2, pp. W24-W28, 2011.
- R. Potestio, T. Aleksiev, F. Pontiggia, S. Cozzini and C. Micheletti, "ALADYN: a web server for aligning proteins by matching their large-scale motion," Nucleic Acids Res, vol. 38(Web Server issue), W41-5, 2010.
- Y. Zhang and J. Skolnick, "TM-align: A protein structure alignment algorithm based on TM-score," Nucleic Acids Res, vol. 33, pp. 2302-2309, 2005.
- I. N. Shindyalov and P. E. Bourne, "Protein structure alignment by incremental combinatorial extension (CE) of the optimal path," Protein Eng, vol. 11, pp. 739–747, 1998.
- L. Holm and C. Sander, "Dali: a network tool for protein structure comparison," Trends Biochem Sci, vol. 20, No. 11, pp. 478–80, 1995.
- T. Madej, C. J. Lanczycki, D. Zhang, P. A. Thiessen, R. C. Geer, A. Marchler-Bauer and S. H. Bryant, "MMDB and VAST+: tracking structural similarities between macromolecular complexes," Nucleic Acids Res, vol. 42, pp. D297-303, 2014).
- J. Razmara, S. Deris and S. Parvizpour, "TS-AMIR: a topology string alignment method for intensive rapid protein structure comparison," Algorithm Mol Biol, vol. 7, 4, 2012.
- Z. H. Zhang, K. Bharatham, W. A. Sherman and I. Mihalek, "deconSTRUCT: general purpose protein database search on the substructure level," Nucleic Acids Res, vol. 38(Web Server issue), W590-W594, 2010.
- Z. H. Zhang, H. K. Lee and I. Mihalek, "Reduced representation of protein structure: implications on efficiency and scope of detection of structural similarity," BMC Bioinformatics, vol. 11, 155, 2010.
- S. Shi, B. Chitturi and N. V. Grishin, "ProSMoS server: a pattern-based search using interaction matrix representation of protein structures," Nucleic Acids Res, vol. 37(Web Server issue), W526-31, 2009.
- C. A. Orengo and W. R. Taylor, "SSAP: sequential structure alignment program for protein structure comparison," Methods Enzymol, vol. 266, pp. 617–635, 1996.
- J. F. Gibrat, T. Madej and S. H. Bryant, "Surprising similarities in structure comparison," Curr Opin Struct Biol, vol. 6, pp. 377-85, 1996.
- G. Mayr, F. Domingues and P. Lackner, "Comparative analysis of protein structure alignments," BMC Struct Biol, vol. 7, 50, 2007.
- L. Holm and P. Rosenström, "Dali server: conservation mapping in 3D," Nucleic Acids Res, vol. 38, pp. W545-549, 2010.
- A. Prlic, S. Bliven, P. W. Rose, W. F. Bluhm, C. Bizon, A. Godzik, P. E. Bourne, "Pre-calculated protein structure alignments at the RCSB PDB website," Bioinformatics, vol. 26, pp. 2983-5, 2010.
- L. Bartoli, E. Capriotti, P. Fariselli, P. L. Martelli and R. Casadio, "The pros and cons of predicting protein contact maps," in: Protein Structure Prediction, M. J. Zaki and C. Bystroff, Eds. 2nd ed., Totowa, New Jersey: Humana Press, 2008.
- J. Hu, X. Shen, Y. Shao, C. Bystroff and M. J. Zaki, "Mining protein contact maps," in Proceedings of BIOKDD02: Workshop on Data Mining in Bioinformatics, with SIGKDD02 Conference, M. Zaki, J. Wang and H. Toivonen, Eds. Edmonton, Alberta, Canada, 2002.
- D. E. Pires, R. C. Melo-Minardi, M. A. Santos, C. H. Silveira, M. M. Santoro and W. Meira, "Cutoff Scanning Matrix (CSM): structural classification and function prediction by protein inter-residue distance patterns," BMC Genomics, vol. 12, S12, 2011.
- D. Bhavani, K. Suvarnavani and S. Sinha, "Mining of protein contact maps for protein fold prediction," Wiley Int. Review on Data Mining and Knowledge Discovery, vol. 1, No. 4, pp. 362–368, 2011.
- H. A. Maghawry, M. G. Mostafa and T. F. Gharib, "A new protein structure representation for efficient protein function prediction," J Comput Biol, vol. 21, pp. 936-46, 2014.
- K. Marsolo and K. Ramamohanarao, "Structure based querying of proteins using wavelets," Proceedings of the 15th ACM International Conference on Information and Knowledge Management (CIKM '06.), November 5–11; Arlington, VA, USA. USA: ACM New York, 2006.
- G. Mirceva, I. Cingovska, Z. Dimov and D. Davcev, "Efficient approaches for retrieving protein tertiary structures," IEEE Trans on Computational Biology and Bioinformatics, vol. 9, No. 4, pp. 1166–1179, 2012.
- B. J. Polacco and P. C. Babbitt "Automated discovery of 3D motifs for protein function annotation," Bioinformatics, vol. 22, pp. 723–730, 2006.
- J. Yang, A. Roy and Y. Zhang, "BioLiP: a semi-manually curated database for biologically relevant ligand-protein interactions," Nucleic Acids Res, vol. 41, D1096-D1103, 2013.
- J. Konc and D. Janezic, "ProBiS–2012: web server and web services for detection of structurally similar binding sites in proteins," Nucleic Acids Res, vol. 40, pp. W214-W221, 2012.
- J. Dundas, Z. Ouyang, J. Tseng, A. Binkowski, Y. Turpaz and J. Liang, "CASTp: computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues," Nucleic Acids Res, vol. 34, W116-W118, 2006.
- N. Furnham, G. L. Holliday, T. A. de Beer, J. O. Jacobsen, W. R. Pearson and J. M. Thornton, "The Catalytic Site Atlas 2.0: cataloging catalytic sites and residues identified in enzymes," Nucleic Acids Res, vol. 42(Database issue), D485-9, 2014.
- K. Kinoshita and H. Nakamura, "Identification of the ligand binding sites on the molecular surface of proteins," Protein Sci, vol. 14, pp. 711-718, 2005.
- Y. Jia, J. Huan, V. Buhr, J. Zhang and L. N. Carayannopoulos, "Towards comprehensive structural motif mining for better fold annotation in the "twilight zone" of sequence dissimilarity," BMC Bioinformatics, vol. 10, No. 1, S46, 2009.
- S. Ku and Y. Hu, "Structural alphabet motif discovery and a structural motif database," Comput Biol Med, vol. 42, pp. 93–105, 2012.
- J. Shi and Y. Zhang, "Fast SCOP classification of structural class and fold using secondary structure mining in distance matrix" in Proceedings of fourth IAPR International Conference (PRIB 2009), V. Kadirkamanathan, G. Sanguinetti, M. Girolami, M., Niranjan and J. Noirel, Eds. September 7–9; Sheffield, UK. Heidelberg: Springer, pp. 344–353, 2009.
- A. Rahimi, A. Madadkar-Sobhani, R. Touserkani and B. Goliaei, "Efficacy of function specific 3D-motifs in enzyme classification according to their EC-numbers," J Theor Biol, vol. 336, pp. 36–43, 2013.
- D.R. Livesay, D. KC and D. La, "Predicting protein functional sites with phylogenetic motifs: past, present and beyond," in Protein Function Prediction for Omics Era, D. Kihara, Ed. USA: Springer, 2011.
- D. KC and D. R. Livesay, "A spectrum of phylogenetic-based approaches for predicting protein functional sites," in Bioinformatics for Systems Biology. S. Krawetz, Ed. New York: Humana Press, 2009.
- B. Nisius, F. Sha and H. Gohlke, "Structure-based computational analysis of protein binding sites for function and druggability prediction," J Biotechnol, vol. 159, No. 3, pp. 123–134, 2012.
- M. N. Wass, L. A. Kelley and M. J. Sternberg, "3DLigandSite: predicting ligand-binding sites using similar structures," Nucleic Acids Res, vol. 38, W469-73, 2010.
- N. Nadzirin, E. Gardiner, P. Willett, P. J. Artymiuk and M. Firdaus-Raih, "SPRITE and ASSAM: web servers for side chain 3D-motif searching in protein structures," Nucleic Acids Res, vol. 40(Web Server issue), W380-6, 2012.
- L. Xie and P. E. Bourne, "A robust and efficient algorithm for the shape description of protein structures and its application in predicting ligand binding sites," BMC Bioinformatics, vol. 8(Suppl 4):S9, 2007.
- L. Sael, D. La, B. Li, R. Rustamov and D. Kihara, "Rapid comparison of properties on protein surface," Proteins, vol. 73, pp. 1–10, 2008.
- L. Sael, B. Li, D. La, et al., "Fast protein tertiary structure retrieval based on global surface shape similarity," Proteins, vol. 72, pp. 1259–1273, 2008.
- D. Kihara, L. Sael, R. Chikhi and J. Esquivel-Rodriguez, "Molecular surface representation using 3d Zernike descriptors for protein shape comparison and docking," Curr Protein Pept Sc, vol. 12, pp. 520–530, 2011.
- R. Chikhi, L. Sael and D. Kihara, "Protein binding ligand prediction using moments-based methods," in Protein Function Prediction for Omics Era, D. Kihara, Ed. USA: Springer, 2011.
- H. Zhao, Y. Yang and Y. Zhou, "Structure-based prediction of DNA-binding proteins by structural alignment and a volume-fraction corrected DFIRE-based energy function," Bioinformatics, vol. 26, pp. 1857–1863, 2010.
- M. E. Bock, C. Garutti and C. Guerra, "Discovery of similar regions on protein surfaces," J Comput Biol, vol. 14, No. 3, pp. 285–99, 2007.
