Технологии высокопроизводительного анализа метилирования ДНК: от генома к панелям генов

Автор: Зуев А.С., Бокова У.А., Васильев С.А.
Журнал: Сибирский онкологический журнал @siboncoj
Рубрика: Обзоры
Статья в выпуске: 6 т.24, 2025 года.
Бесплатный доступ
Актуальность. Метилирование ДНК регулирует множество биологических процессов, опосредуя нормальное развитие организма. Нарушения в паттернах метилирования ассоциированы с многочисленными патологическими состояниями, в особенности с наследственными и онкологическими заболеваниями. Важность данных аномалий подчеркивается их активным использованием в качестве клинически значимых биомаркеров для стратификации пациентов, мониторинга течения болезни, ранней диагностики и прогноза ответа на терапию. Выявление специфических паттернов метилирования возможно с помощью таргетных высокопроизводительных методов, обеспечивающих фокус на ключевых регионах интереса. Цель исследования – анализ и обобщение литературных данных, описывающих применение технологий высокопроизводительного анализа метилирования ДНК, в том числе технологий, основанных на целевых (таргетных) подходах. Материал и методы. Проведен систематический анализ литературных данных по базам данных PubMed, Web of Science, Scopus, посвященных особенностям проведения высокопроизводительного анализа метилирования ДНК при онкологических и некоторых генетически обусловленных патологиях. Проанализировано 113 источников, охватывающих период с 2000 по июнь 2025 г., 32 из которых использованы для написания обзора. Результаты. Обобщены сведения о существующих технологиях высокопроизводительного анализа метилома, методах конверсии ДНК, их преимуществах и ограничениях. Рассмотрены существующие методы таргетного обогащения, их сильные и слабые стороны, а также возможности применения в научной и диагностической практике. Заключение. Определение статуса метилирования ДНК перестало быть инструментом фундаментальных исследований, став одной из важных областей трансляционной медицины, особенно в онкологии. Современные методы анализа метилома позволяют выявлять эпигенетические маркеры для диагностики и прогноза заболеваний, выбирать оптимальную терапию, проводить поиск молекулярных мишеней для таргетных препаратов. Целевое обогащение ДНК позволяет повысить точность и чувствительность, снижая при этом стоимость анализа, а применение некоторых подходов способствует приложению таргетного анализа к сложным образцам. В совокупности с гибкостью выбора регионов интереса такие качества обусловливают применение целевых подходов не только в научно-исследовательской, но и в практической деятельности.
Метилирование ДНК, эпигенетика, секвенирование, диагностика
Короткий адрес: https://sciup.org/140313334
IDR: 140313334 | УДК: 577.213.32:575:616-006 | DOI: 10.21294/1814-4861-2025-24-6-149-159
High-throughput DNA methylation analysis technologies: from genome to gene panels
Background. DNA methylation regulates numerous biological processes, mediating normal development. Alterations in methylation patterns are associated with multiple pathological conditions like hereditary diseases and cancer, making them valuable clinical biomarkers for patient stratification, disease monitoring, early diagnosis, and prediction of response to therapy. Highly targeted, high-throughput methodologies focusing on critical genomic loci enable precise identification of distinct methylation signatures. the aim of the study was to analyze and summarize literature data describing the use of high-throughput DNA methylation analysis technologies, including those based on targeted approaches. Material and Methods. A systematic analysis of literature data was conducted using the PubMed, Web of Science, and Scopus databases, focusing on the characteristics of high-throughput DNA methylation analysis used in cancer and some genetic diseases. A total of 113 sources were analyzed, chronologically covering the period from 2000 to June 2025, 32 of which were used to write the review. Results. The existing technologies for high-throughput methylome analysis, DNA conversion methods, and their advantages and limitations were summarized. In addition, the current targeted enrichment methods, their strengths and weaknesses, and potential applications in scientific and diagnostic practice were discussed. Conclusion. DNA methylation analysis has evolved from a basic research tool into a cornerstone of translational medicine, particularly in oncology. Modern methylome analysis techniques facilitate the discovery of epigenetic markers critical for diagnosing diseases, assessing prognosis, guiding therapy selection, and identifying molecular targets for targeted drugs. Targeted DNA enrichment increases analytical precision and sensitivity while reducing costs. Furthermore, specialized strategies permit targeted analysis even with challenging samples. Combined with the flexibility to focus on specific genomic regions, these advantages make targeted approaches viable not only in academic research but also in routine clinical diagnostics.
Текст научной статьи Технологии высокопроизводительного анализа метилирования ДНК: от генома к панелям генов
Метилирование ДНК – эпигенетический механизм, играющий ключевую роль в регуляции экспрессии генов, поддержании стабильности генома, клеточной репликации и дифференцировке. Наиболее изученной модификацией ДНК у человека является метилирование цитозина в составе CpG-динуклеотидов в С5 позиции цитозинового кольца с образованием 5-метилцитозина (5mC). Присоединение метильной группы (-CH₃) к цитозину происходит при помощи ДНК-метилтрансфераз (DNMT1, DNMT3a и DNMT3b), что приводит к изменению структуры хроматина и, как следствие, к подавлению или активации транскрипции генов. Производное 5mC – 5-гидроксиметилцитозин (5hmC) – образуется при окислении метильной группы и добавлении к ней гидроксильной группы и также считается как самостоятельным эпигенетическим маркером, ассоциированным с активной транскрипцией и регуляцией альтернативного сплайсинга, так и промежуточной стадией активного деметилирования. Эти модифицированные основания являются наиболее распространенными эпигенетическими маркерами, обеспечивающими контроль над матричными процессами, онтогенезом, инактивацией X-хромосомы, геномным импринтингом и подавлением транспозонов, а их нарушения ассоциированы с широким спектром патологий, включая онкологические и нейродеге-неративные заболевания, болезни импринтинга и другие [1].
Эпигенетические нарушения, в частности аномалии метилирования ДНК, играют критическую роль в патогенезе ряда заболеваний. При онкологических заболеваниях дисбаланс метилирования проявляется в двух основных формах. С одной стороны, наблюдается гиперметилирование промо-торных регионов генов-супрессоров опухолевого роста, что приводит к их ингибированию. К числу таких генов относятся регуляторы клеточного цикла (RB1, CDKN2A, CDKN2B, TP53, BRCA1) [2, 3], основной компонент апоптосомы APAF1 [4], а также ген MLH1, ответственный за репарацию неспаренных оснований ДНК [5]. С другой стороны, глобальное гипометилирование генома способствует онкогенезу за счет реактивации онкогенов и мобильных генетических элементов, а также индукции хромосомной нестабильности [6, 7]. При нейродегенеративных патологиях, таких как болезни Паркинсона и Альцгеймера, аномалии метилирования выявлены в генах тау-белка (MAPT), α-синуклеина (SNCA), пресенилина (PSEN1) и других ключевых белков, что указывает на глубокую эпигенетическую составляющую в механизмах развития этих заболеваний [8–10]. Отдельную категорию составляют болезни импринтинга, этиология которых напрямую обусловлена аллель-специфичными нарушениями метилирования в локусах центров импринтинга. Данные нарушения, затрагивающие отцовский или материнский аллель, вызывают дисбаланс в экспрессии импринтированных генов, что, в свою очередь, клинически манифестирует в виде специфических наследственных синдромов [11].
Установленная функциональная связь между развитием патологических состояний и аберрантным метилированием ДНК определяет его значимость в качестве высокоспецифичного диагностического маркера. Его использование позволяет проводить верификацию диагноза в сложных случаях. Всестороннее изучение роли эпигенетических механизмов является необходимым условием для углубления понимания молекулярных основ заболеваний и в долгосрочной перспективе служит фундаментом для разработки инновационных методов терапии, направленных на коррекцию эпигенома. Важную роль в этом процессе играют технологии таргетного анализа, обеспечивающие целенаправленное исследование профиля метилирования. Универсальность данных методов обусловливает их широкое применение как в области фундаментальных научных исследований, так и в практической клинической диагностике.
Целью исследования являются систематизация и анализ современных методов таргетного обогащения, предназначенных для анализа метилирования ДНК, с оценкой их потенциала и ограничений.
Методы высокопроизводительного анализа метилирования ДНК
В настоящее время разработан широкий спектр методов, позволяющих исследовать метилом как глобально на уровне всего генома, так и таргетно на уровне конкретного локуса с однонуклеотидным разрешением. Современные, наиболее часто применяемые генетические подходы включают полногеномное бисульфитное секвенирование (Whole Genome Bisulfite Sequencing, WGBS), бисульфитное секвенирование ограниченного набора локусов (Reduced representation bisulfite sequencing, RRBS), селективные генетические панели и метилочипы, каждый из которых обладает уникальными преимуществами и ограничениями.
WGBS – высокопроизводительный метод, обеспечивающий наиболее полный анализ паттернов метилирования с однонуклеотидным разрешением по всему геному. Основанный на бисульфитной конверсии ДНК с дальнейшим массовым параллельным секвенированием и биоинформатической обработкой, WGBS позволяет проанализировать метилирование в CpG и не-CpG сайтах. WGBS является наиболее полным и точным методом анализа метилирования ДНК, позволяющим получать детальные эпигенетические данные для фундаментальных исследований [12] и клинической практики, например для разработки высокоспецифичных и чувствительных биомаркеров для ранней диагностики и типирования опухолей [13]. Однако большой объем получаемых данных при WGBS приводит к высокой стоимости и сложности биоинформатической обработки.
Главным путем снижения стоимости анализа является уменьшение количества анализируемых CpG-сайтов в геноме путем обогащения отдельных регионов генома в анализируемом образце ДНК. Одним из подходов к такому обогащению является метод RRBS. Основанное на тех же принципах, что и полногеномное секвенирование, RRBS имеет этап энзиматической фрагментации, ограничивающий анализируемые регионы генома CpG-богатыми сайтами. Высокое покрытие таких регионов в совокупности с охватом до 80 % промоторов и регуляторных элементов (от 2 до 7 млн CpG-сайтов) снижает также вычислительные затраты, что особенно актуально для больших популяционных исследований [14].
Другим способом анализа только части CpG-сайтов в геноме является метод, использующий микрочипы от компании Illumina. Изначально создаваемые для онкологических задач микрочипы позволяли одновременно анализировать большое количество CpG-сайтов, что было реализовано в создании метилочипа «GoldenGate Methylation Cancer Panel I», включавшего 1505 CpG-сайт 807 генов, преимущественно известных супрессоров и онкогенов. Последней версией метилочипов является Infinium MethylationEPIC (850K), позволяющая проанализировать около 850 000 CpG-сайтов, покрывая при этом около 99 % генных промоторов. Технология микрочипов является наиболее проверенным методом исследования метилирования ДНК, что делает ее стандартом для сравнения новых технологий. Главным ограничением данного метода является доступность микрочипов преимущественно для человека и невозможность анализа других видов. Кроме того, микрочипы имеют ограничения в анализе повторяющихся элементов и редких аллелей.
Более таргетными и гибкими высокопроизводительными инструментами являются целевые панели, которые, используя гибридизационный захват и бисульфитное секвенирование регионов, могут обеспечивать более высокие целевые по-
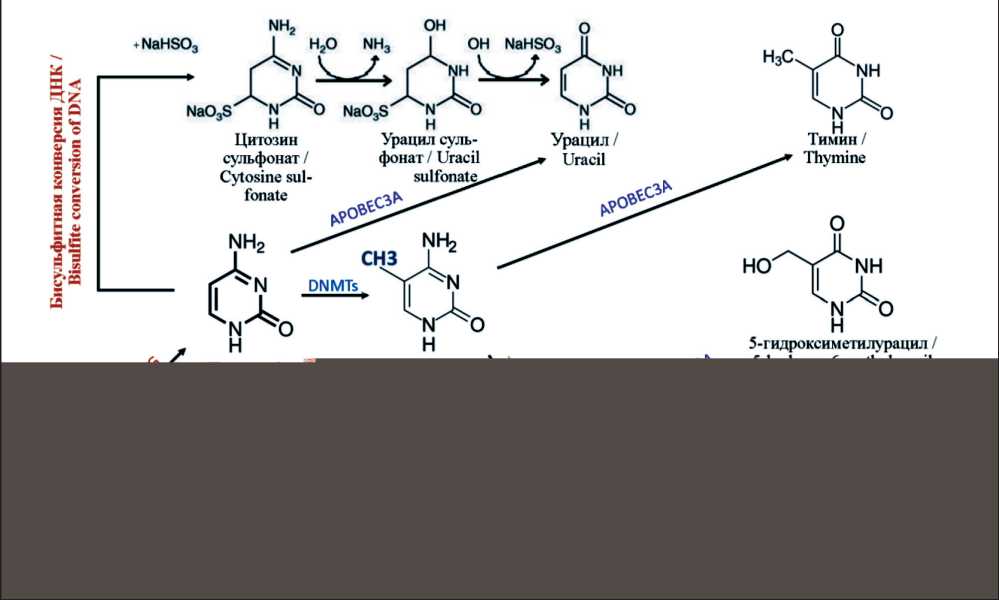
Рис. 1. Возможные биохимические модификации цитозина и точки приложения ферментов и химических модификаторов конверсии ДНК. DNMTs-ДНК-метилтрансферазы (DNMT1, DNMT3a и DNMT3b), TETs-Tet-метилцитозиндиоксигеназы, APOBEC3A-каталитическая субъединица 3A редактирования мРНК аполипопротеина B, T4-BGT-бета-глюкозилтрансфераза фага T4, TDG-тиминовая гликозилаза. Примечание: рисунок выполнен авторами
Fig. 1. Possible biochemical modifications of cytosine and the application points of enzymes and chemical modifiers of DNA conversion. DNMTs-DNA methyltransferases (DNMT1, DNMT3a, and DNMT3b), TETs-Tet-methylcytosine dioxygenases, APOBEC3A-the catalytic subunit 3A of apolipoprotein B mRNA editing, T4-BGT-phage T4 beta-glucosyltransferase, TDG-thymine glycosylase.
Note: created by the authors
казатели глубины покрытия при вариабельном охвате. Технология гибридизационного захвата обеспечивает высокую специфичность отбора регионов интереса, что дает возможность анализа редких аллелей. Размерность таргетных панелей может варьировать от анализа более 3,5 млн CpG-сайтов с помощью панели Twist Human Methylome (Twist Bioscience) до анализа конкретных регионов и сайтов, например, применяемых для подтверждения данных полнометиломных исследований. Таргетные эпигенетические панели отличаются информативностью, экономичностью и возможностью индивидуализации, что позволяет применять их под конкретные научные и диагностические задачи. Анализ состояния метилирования конкретных генов, их регуляторных элементов позволяет минимизировать биоинформатическую обработку и сократить время диагностического поиска.
Общим этапом в вышеописанных методах является бисульфитная модификация ДНК. Обработка ДНК бисульфитом натрия преобразует не-метилированные цитозины в урацил, не подвергая этой реакции цитозины, содержащие метильную группу (рис. 1). После бисульфитной обработки ДНК подвергается ПЦР-амплификации, в процессе которой урацил в ампликонах заменяется на тимин, а метилированные цитозины остаются цитозинами. Использование бисульфитной обработки связано с рядом ограничений, такими как деградация ДНК вследствие химического воздействия, появление артефактов амплификации из-за наличия урацила в составе ДНК, отсутствие различий в конверсии 5mC и гидроксиметилцитозина (5hmC), а также с возможностью неполной конверсии ДНК. Для преодоления некоторых из этих ограничений в последние годы предложено несколько способов анализа уровня метилирования без использования бисульфитной модификации, в частности применение энзиматической конверсии ДНК.
В клетке существует механизм перевода метилированного цитозина в неметилированное состояние с использованием фермента TET (Ten-eleven translocation) через промежуточные продукты: 5hmC, формилцитозин (5fC) и карбоксицитозин (5caC) (рис. 1). Рекомбинантный TET используется в TET-ассистированном пиридинборановом секвенировании (IrTAPS) [14]. Окисление 5mC и 5hmC до 5caC и последующее восстановление до дигидроурацила позволяют распознавать последнюю модификацию цитозина как тимин при секвенировании. Такой подход нивелирует био-информатическую коррекцию, необходимую при бисульфитной конверсии, а сохранение длины молекулы ДНК более 10 т.п.н. позволяет использовать технологии, основанные на длинных прочтениях (Nanopore, PacBio) и применяющиеся для фазирования эпигенома, то есть привязки информации о метилировании цитозина только к одной из пары гомологичных хромосом [15]. Получение данных фазирования может дополнить понимание функций аллель-специфических эпигенетических модификаций при различных патологических состояниях. Возможным ограничением метода является зависимость эффективности и специфичности окисления белками TET от доступности региона хроматина, а также суммарный анализ различных модификаций цитозина.
Сходным методом детекции модификаций цитозина является ферментативное метил-секвенирование ( enzymatic methyl-seq, EM-seq ). Данный метод основан на действии трех ферментов: Tet-метилцитозиндиоксигеназы 2 (TET2), катализирующей окисление 5mC до 5hmC, затем 5fC и, наконец, 5caC, бета-глюкозилтрансферазы фага T4 (T4-BGT), катализирующей глюкозили-рование как образованного TET2, так и геномного 5hmC до 5-β-глюкозилоксиметилцитозина (5gmC) и каталитической субъединицы 3A редактирования мРНК аполипопротеина B (APOBEC3A), дезаминирующей цитозины, но не модифицированные формы 5mC или 5hmC [16] (рис. 1). Такой подход, как и при IrTAPS, способствует сохранению длинных фрагментов ДНК, имея лучшие показатели качества библиотек в сравнении с бисульфитным секвенированием. Сохранение таких преимуществ при использовании малых количеств ДНК позволяет использовать подход ферментативного метилирования как в исследовательских, так и в диагностических целях при анализе единичных клеток или внеклеточной ДНК для раннего выявления гепатоцеллюлярной карциномы [17].
Наконец, развитие методов нанопорового секвенирования отдельных молекул сделало возможной прямую детекцию метилирования ДНК без предварительной бисульфитной или ферментативной модификации цитозина. При нанопоровом секвенировании нативная ДНК проходит через белковый канал (нанопору), где каждое азотистое основание создает уникальный сигнал. Различия сигнатур сигнала для различных модификаций нуклеотидов позволяют при анализе отличать 5mC, 5hmC и другие эпигенетические модификации. При этом нанопоровое секвенирование позволяет избежать процессов модификации ДНК и ПЦР-амплификации, что нивелирует появление артефактов этих этапов при анализе данных. Длина прочтений до нескольких десятков тысяч оснований позволяет получить информацию об аллель-специфичном метилировании ДНК, давая возможность обнаружения новых импринтиро-ванных регионов и регуляторных элементов, в том числе в контексте межклеточной и межтканевой гетерогенности.
Анализ метилома единичных клеток
Анализ метилома единичных клеток является перспективным направлением эпигенетики, обусловленным как фундаментальными биологическими вопросами, такими как гетерогенность, развитие и дифференцировка клеток, так и практическим приложением в медицине. Для построения геномных карт метилирования ДНК с разрешением на уровне отдельных нуклеотидов в единичных клетках используется метод single-cell Bisulfite Sequencing (scBS-seq) [18]. Основанный на бисульфитной конверсии ДНК и постбисуль-фитном присоединении адаптеров (post-bisulfite adaptor tagging, PBAT), такой подход позволяет ограничить потерю информативных последовательностей, которая обычно происходит при обработке бисульфитом библиотек с адаптерами. Дальнейшая преамплификация с вырожденными гексапраймерами позволяет усилить сигнал при малом количестве ДНК, давая возможность проводить анализ на отдельных клетках. Такой подход хоть и обеспечивает высокие показатели глубины секвенирования и покрытия CpG-сайтов при относительно малом количестве исследуемой ДНК, но не позволяет избежать артефактов бисульфитной конверсии и ПЦР, таких как укорочение фрагментов и смещение преимущественно в сторону GC-богатых регионов вследствие амплификации с вырожденными праймерами, а также не позволяет различить 5mC и 5hmC при последующей биоин-форматической обработке.
Еще один подход анализа единичных клеток представлен технологией одноядерного метиломного секвенирования (snmC-seq3). Этот метод высокопроизводительного анализа метилома объединяет выделение и сортировку ядер, би-сульфитную конверсию ДНК, высокоэффективное мультиплексное ПЦР-обогащение и секвенирование с помощью коротких прочтений. Особенностью метода стала минимизация технических артефактов, позволившая увеличить покрытие и глубину секвенирования CpG и снизить шумы конверсии в сравнении с предыдущими итерациями метода, а интеграция snmC-seq3 с транс-криптомными данными и анализом конформации хроматина позволяет отнести метод в область пространственной геномики [19]. Позволяя изучить эпигенетическое разнообразие с относительно высоким разрешением, применение snmC-seq3 кажется особенно ценным в нейробиологии и онкологии, где клеточная гетерогенность определяет патогенез болезней.
Одновременное профилирование транскрип-тома, метилома ДНК и доступности хроматина в одной клетке было показано ранее с помощью метода scNMT-seq (секвенирование нуклеосом отдельных клеток, метилирование и транскрипция) [20]. Получая данные об экспрессии генов, метилировании ДНК и распределении GpC-меток во вненуклеосомных участках ДНК, scNMT-seq позволяет напрямую выявлять зависимость между метилированием ДНК, доступностью хроматина и экспрессией генов в отдельных клетках. Несмотря на 50 % потерю покрытия CpG вследствие фильтрации сайтов G-C-G и C-C-G, техническую сложность протокола и требования глубокого секвенирования (в среднем 16 млн прочтений на клетку для BS-seq и 2 млн прочтений для RNA-seq), scNMT-seq показал высокое разрешение и покрытие по доступности хроматина (~85 % тел генов и ~75 % промоторов) и метилому (более 1 млн CpG-сайтов, охватывающих ключевые регуляторные последовательности, после фильтрации артефактных позиций), позволяя по данным о доступности GpC-сайтов реконструировать позиции нуклеосом с шагом ~200 п.н. [20].
Применение методов интегративного анализа эпигенетики и транскриптомики предоставляет беспрецедентные возможности для изучения регуляции генов, дифференцировки и гетерогенности клеточных популяций. Совмещая в себе различные типы данных, такие методы способны раскрывать причинно-следственные связи между уровнями реализации генетической информации, что недостижимо при раздельном анализе.
Таким образом, возможности анализа метилирования ДНК предоставлены широким спектром современных методов, каждый из которых имеет свои преимущества и ограничения (табл. 1). Развитие отдельных этапов позволяет преодолевать некоторые недостатки методов, что способствует их применению для более глубокого анализа эпигенома. Примерами таких достижений может быть уже описанная технология постбисульфитного мечения адаптерами, снижающая потери участков ДНК и увеличивающая длину вставки и скорость картирования прочтений [21], или технологии энзиматической конверсии, способствующие более полному преобразованию модифицированных оснований и сохраняющие длинные фрагменты ДНК. Отдельно стоит отметить развитие на-нопорового секвенирования, точность которого изначально составляла около 70 – 80 %, но с совершенствованием технологий достигла уровня методов массового параллельного секвенирования [22], обладая преимуществом в сборке геномов и фазировании генотипов. Несмотря на значительные преимущества, ключевым ограничением всех методов анализа геномного профиля метилирования является их высокая стоимость. В связи с этим для глубокого анализа конкретных геномных регионов необходимо таргетное обогащение.
Методы таргетного обогащения
Методы таргетного обогащения предназначены для анализа специфических участков ДНК, в том числе регионов, подвергающихся метилированию, и сочетают в себе высокую точность с экономической эффективностью, что делает их идеально подходящими для прикладных исследований и клинической диагностики. Наиболее часто применяе- мые технологии таргетного обогащения включают гибридизационное или ПЦР-обогащение [23].
При гибридизационном обогащении используются одноцепочечные олигонуклеотидные зонды для таргетного захвата специфических участков ДНК. Гибридизация может происходить с зондами, иммобилизированными на твердой поверхности (например, на магнитных частицах или чипе), или при взаимодействии последовательностей нуклеиновых кислот и зондов в растворе. Данный тип обогащения зависит от параметров длительности гибридизации, размеров зондов, GC-состава, но возможность одновременного анализа сотен регионов в одном запуске позволяет использовать гибридизационное обогащение в масштабных поисковых исследованиях генома [23, 24].
Обогащение с помощью амплификации происходит с использованием олигонуклеотидов, ограничивающих регионы интереса и позволяющих кратно увеличить их копийность с помощью ПЦР. Такой метод лучше подходит при работе с малым количеством ДНК и при низком качестве исходной матрицы. Преимущества ПЦР-обогащения (быстрота постановки реакции, гибкость выбора регионов для анализа и экономическая эффективность) позволяют применять метод при многих типах диагностических (например, для исследования на мутации в генах BRCA1/2 ) и исследовательских задач (подтверждение результатов полногеномных подходов). Разработка мультиплексной панели ПЦР-обогащения требует одновременной работы от десятков до сотен различных праймеров, условия для которых оптимизируются усреднением, что приводит к ограничению отбора возможного количества регионов интереса для определенной панели. Ограниченное количество регионов тем не менее может применяться в диагностических «скрининговых» целях, где одной из основных задач является быстрый целевой поиск причины патологии. Применение такого метода уже нашло отражение в диагностической панели для расстройств аутистического спектра (панель Яско), выявляющей нарушения в генах, вовлеченных в регуляцию метилирования ДНК [23, 25].
Одним из достижений методов ПЦР-обогащения может считаться амплификация с уникальными молекулярными индексами (unique molecular identifiers, UMIs). Технология UMIs устраняет ошибки ПЦР путем маркировки до амплификации каждой молекулы нуклеиновой кислоты уникальными последовательностями. Такая маркировка помогает отличить вариантные аллели, присутствующие в исходном образце, от ошибок, допущенных при подготовке библиотеки, обогащении или секвенировании. UMIs позволяют контролировать смещения амплификации и появление дупликатов, связанные с подготовкой образцов методом ПЦР, а также количественно определить абсолютное число молекул без необходимости детектировать
Таблица 1/table 1
w
> го с го с о го
> -С ф Е < Z
о
3 о.
-С О)
о
О)
р
О) р о с
-С о ф



8 й
3 О

х
й

s S
й N
к
н
к
S
н


о
8 о й 0) ад
Note: created by the authors.
каждую отдельную молекулу или определять количество ее копий. Применение UMIs было показано при обнаружении гиперметилированных CpG-островков в циркулирующей внеклеточной ДНК пациентов с гепатоцеллюлярной карциномой [26]. Такой подход обеспечил не только минимизацию технических шумов ПЦР, но и повысил точность количественного анализа метилирования и чувствительность определения малого количества ДНК, необходимых для диагностики ранних стадий гепатоцеллюлярной карциномы по циркулирующей внеклеточной ДНК.
Обогащение для анализа метилома может проводиться энзиматически, с помощью ферментов рестрикции. Такой метод основан на избирательной фрагментации ДНК по сайтам, ограничивающим определенные регионы интереса. Обогащение и отбор целевых регионов позволяют проводить анализ с различной степенью охвата генома, сохраняя при этом возможность оптимизации за счет комбинации рестриктаз [27, 28].
Для обогащения целевых участков генома также могут применяться методы, основанные на CRISPR-Cas9 технологии. Основные этапы CRISPR-Cas9-обогащения включают избирательное связывание неактивной формы белка Cas9 (dCas9) с целевыми регионами генома посредством направляющих РНК (sgRNA). Дальнейшая иммунопреципитация или использование маг-нитных/стрептавидиновых шариков позволяет отбирать фрагменты с высокой эффективностью для уникальных локусов, в том числе избавляя от артефактов секвенирования за счет отсутствия этапа амплификации [29]. Одним из методов, основанных на CRISPR-Cas9-обогащении, является CRISPR-Cap, где применяется технология отрицательного обогащения Nanopore Cas9-targeted sequencing (nCATS). При этом методе используется система биотин-стрептавидинового захвата фрагментов ДНК, 3’ и 5’ концы которых расщепляются комплексами Cas9/sgRNA. Затем экзонуклеаза переваривает неспецифические фрагменты, при этом интересующая область защищается Cas9/sgRNA [30]. Увеличивая таким образом пропускную способность и специфичность обогащения с возможностью отбора мишеней до 200 т.п.о. и оценки частоты редких аллелей до 1 %, одновременно снижая вероятность возникновения артефактов и затраты секвенирования, CRISPR-Cas9-обогащение является методом, приложение которого можно найти в диагностике эпигенетического (как в случае количественной оценки метилирования гена MGMT в клеточных линиях человека и диффузной глиоме [30]) и генетического статуса (например, для секвенирования гена рака молочной железы BRCA1 с помощью нанопорового секвенирования [31]) при различных патологических состояниях.
Таргетное обогащение может не требовать физического обогащения образцов и выполняться непосредственно во время секвенирования. При нанопоровом секвенировании технология селективного обогащения позволяет отбирать молекулы ДНК по их принадлежности к целевым регионам с помощью вычислительных методов. С помощью картирования начала секвенируемой последовательности ДНК на референсный геном биоинфор-матическими алгоритмами в реальном времени принимается решение о продолжении секвенирования при принадлежности участка к региону интереса или прекращении чтения последовательности при невозможности картирования на таргетный регион [32]. Потенциально такое обогащение может использоваться при анализе метилома, в том числе аллель-специфического определения статуса метилирования при патологиях, но будет иметь прямую зависимость эффективности от доступных вычислительных мощностей.
Как и любые другие методы, методы таргетного обогащения имеют свои преимущества и недостатки (табл. 2), однако основной их задачей является отбор целевых участков генома с дальнейшим применением высокопроизводительных методов и анализом полученных результатов. В настоящее время с помощью методов таргетного обогащения создано множество индивидуальных панелей под разные нозологии, в том числе в онкологии, медицинской генетике, кардиологии и многих других областях медицины.
Заключение
Метилирование ДНК является основным эпигенетическим механизмом, регулирующим экспрессию генов, а его нарушение связано с возникновением различных типов патологий. Современные технологии анализа метилома неразрывно связаны с методами целевого обогащения ДНК. Развитие этих методов в направлении повышения точности, чувствительности и применимости к сложным клиническим образцам в совокупности направлено на удовлетворение растущих требований клинической диагностики и трансляционных исследований. Обогащение целевых регионов стало эффективным фактором снижения стоимости и продолжительности секвенирования, а возможность работать с преаналитически сложными образцами, такими как циркулирующая бесклеточная ДНК и ДНК из архивного материала, расширяет потенциал практического применения как готовых панелей (особенно в онкологии), так и платформ для создания пользовательских решений. Существующие на данный момент эпигенетические панели показали свою ценность в онкологии, неврологии, генетике и многих других направлениях медицины, позволяя выявлять эпигенетические маркеры для ранней диагностики, прогноза заболеваний, определения чувствительности к терапии. Эпигенетические панели таргетного обогащения занимают свое определенное место в развитии
Таблица 2/table 2
^ X ЕЕ s X 0)
го I— о ю о о
о
0) I— О. го
с 0) Е
£ о
X с 0) < Z о
75 ф ф О) а го
о I— 0)
о
W 75
О £
Ф


Л
S
я о S
я й
Я и
я а я
я
я
й
2 зЯ
я
ЗЯ
Я -
Я s
О к
Я
к
я я 4) 2
я я
£ Я и
и и
аз
я S еб я о я
я S
S
я
g я S о я
я 2 Я § й
5 к я се
я й § g й се S
W й
я
S
2 * и я
О зЯ я 2
я и
К
я о S
к
S о я
й
а
£ й
а
2 Й
Й S
2 .Й
8 S 4) ад
X
а
S я
я я 4) й
й о ад
Й
й '5
к
я
W я
Й
а 8 с 8
я
я
S
й
й
й
2 °
■П й ад
ад й
й
я
й о я
S
2 X
й
й
я й я я я
к
й о S се й
я
я
я я
£
Z
Q
й
§
й
S се
й
й
Я
§ й
я я
W я
)S
S
я
й
н Й
я я
я
S ! а я
й О 8
В о
’8 ’Л ад
5 и
й
я
§
я •8я § S
я й
й
23 3
8 J
Я 'й
ад
й .8
й
£
ад ,й и э и Й о ’ад
й
й
£ ° о
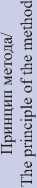

о
ж я Я я X я я я о в
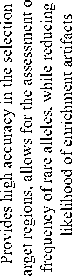

Я
§ 2 Я Я Я S
о
Я й
2 > •
я •;
се
а а
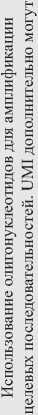
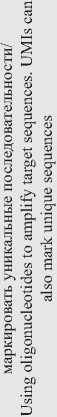

S
Я

зя я я я
Note: created by the authors.
персонализированной и предиктивной медицины, становясь незаменимым инструментом диагностики наряду с геномными и транскриптомными технологиями. При этом интеграция данных подходов показывает важность, актуальность и недостаток понимания вопросов, лежащих в основе
