The effect of evolutionary algorithm in gene subset selection for cancer classification

Author: M.N.F. Fajila, M.A.C. Akmal Jahan
Journal: International Journal of Modern Education and Computer Science @ijmecs
Article in issue: 7 vol.10, 2018.
Free access
The fact that reflects the cancer research consequences shows that still there are improvements that should be investigated in the stream of cancer in future. This leads the researchers to actively involve further in cancer research field. As an invention, a hybrid machine learning method is proposed in this study where two filters are assessed along with a wrapper approach. Typically, filters prioritize the features while, wrappers contribute in subset identification. Though both filters and wrappers exist independently, the excellent results they produce when applied subsequently. The wrapper-filter combination plays a major role in feature selection. Yet, incorporating with a best strategy for feature space analysis is crucial in this concern. Thus, we introduce the Evolutionary Algorithm in the proposed study to search through the feature space for informative gene subset selection. Though there are several gene selection approaches for cancer classification, many of them suffer from law classification accuracy and huge gene subset for prediction. Hence, we propose Evolutionary Algorithm to overcome this problem. The proposed approach is evaluated on five microarray datasets, where three out of them provide 100% accuracy. Regardless the number of genes selected, both filters provide the same performance throughout the datasets used. As a consequence, the Evolutionary Algorithm in feature space search is highlighted for its performance in gene subset selection.
Evolutionary Algorithm, Filters, Gene Subset, Microarray, Wrappers
Short address: https://sciup.org/15016780
IDR: 15016780 | DOI: 10.5815/ijmecs.2018.07.06
Text of the scientific article The effect of evolutionary algorithm in gene subset selection for cancer classification
Published Online July 2018 in MECS DOI: 10.5815/ijmecs.2018.07.06
The uncontrolled growth of cells in the human body is known as cancer. Although there is a huge improvement in medical field today, a complete solution for some diseases seems to be mysterious. Nevertheless, few treatments cure the disease for certain extend when there is a possibility to detect the disease in their early stage and one such disease is cancer. Thus, identification of cancer is a vital problem in current research field.
Alternatively, microarray, a high-throughput technology is prominently used in cancer detection and classification. Although microarray data is rich in information, the volume of the data makes the task of handling it challengeable. Moreover, microarray experiments typically provide large amount of genes relatively number of instances. This may leads to problems such as overfitting, increase in training time and decrease in prediction accuracy. Hence, microarray data analysis is crucial in gaining its valuable knowledge in the field of cancer research. This paper is structured as follows; Section II revises related work; Section III and IV describe methodology and results respectively. The conclusion is summarized in Section V.
-
II. Related Work
Several techniques have been made in microarray analysis and one such technique is Machine Learning. Further, classification and dimensionality reduction are two machine learning techniques. Moreover, feature selection plays a major role in classification of high dimensional data such as microarray data. The microarray data is enriched with irrelevant and redundant features (i.e. genes). Thus, gene selection eliminates these redundant and irrelevant genes making the dimension of the dataset small in size through providing the most informative gene subset.
Several approaches have been already carried out for cancer classification in the past decades. Some methods provide poor performance in terms of time complexity [1, 2] whereas few approaches result in a huge gene subset [3, 4]. Selecting a smallest informative gene subset which can predict an unknown sample perfectly is still challenging. Voting machines and self-organizing maps (SOM) were used in Leukemia subtype classification [5]. Furey et al. [6] and Guyon et al. [7] used Support Vector Machine (SVM) for cancer classification. Further, Wang [8] proposed a correlation-based feature selector evaluated on leukemia and diffuse large B-cell lymphoma datasets. Though diffuse large B-cell lymphoma was classified perfectly, Leukemia was classified with three misclassifications in [8]. A novel Markov blanket-embedded genetic algorithm (MBEGA) was assessed on eleven microarray datasets [9]. Yet, none of the dataset out of eleven was classified with 100% accuracy. Sun [10] applied a genetic algorithm based approach for clustering on an artificial dataset and a breast cancer dataset.
Recently, Bouazza et al. [11] carried out a comparative study on feature subset selection. Five cancer microarray datasets were evaluated using five supervised classifiers. Consequently, they [11] suggested the signal to noise ratio feature selection method with K Nearest Neighbors classifier for feature selection. Comparatively wrappers and filters are used simply with good performance [2, 8, 11-17]. Yet, some studies show lack of performance due to direct application of wrappers into the original datasets [2]. Moreover, the studies show that the classification accuracy highly depends on the type of the filter and as well as the type of the wrapper together with the search strategy [18]. Thus, selecting the most appropriate wrapper and filter seems to be challenging. Further, it seems the Evolutionary Algorithms (EA) [19-27] such a Genetic Algorithm (GA) [19, 20], Particle Swam Optimization Algorithm (PSO) [19, 23], Ant Colony Optimization Algorithm (ACO) [22] and Artificial Bee Colony Algorithm (ABC) [24] have been successfully applied in many researches in gene subset selection. Alba [19] proposed a wrapper approach with EA and SVM namely PSO-SVM and GA-SVM. The approach was evaluated on six microarray datasets achieving good results with few significant genes. Further, Xiong and Wang [22] proposed a gene selection method which in cooperates with ACO and random forest. The efficiency of the proposed approach is reflected when evaluated on Colon cancer dataset and Leukemia dataset. Recently, Alshamlan [24] proposed an algorithm namely, Co-ABC which uses Correlation-based Feature Selection filter for preprocessing and ABC algorithm for gene selection. The approach was evaluated on six microarray datasets out of which five provided 100% accuracy with only few genes. The author compares the results with ABC-SVM [25], minimum Redundancy Maximum Relevance-ABC (mRMR-ABC) [26], Co-GA and Co-PSO algorithms. The results obtained using Co-ABC leads all the algorithms which were compared. Hence, we propose the EA in this study for gene subset selection in cancer classification as shown in Fig. 1.
-
III. Methodology
In this study a new hybrid feature selection approach is employed as described in Fig. 1. The dimensionality of the dataset is reduced initially with a preprocessing step using a filter with a ranker search method. Then the resultant gene subset is further processed with a wrapper approach where the ultimate informative gene subset is resultant. Evolutionary Algorithm is used in wrapper approach to search through the gene space to select the most informative gene subset for the prediction of the test dataset. Typically there are three feature selection methods namely, filters, wrappers and embedded method. Filters are widely used in preprocessing to reduce the dimensionality of the original dataset. Yet, defining a threshold below which the features are eliminated from being ranked is the major problem in the filter approach. One heuristic approach known as n-1 rule where n denotes the number of samples selects the most informative n-1 genes to start the analysis [8].
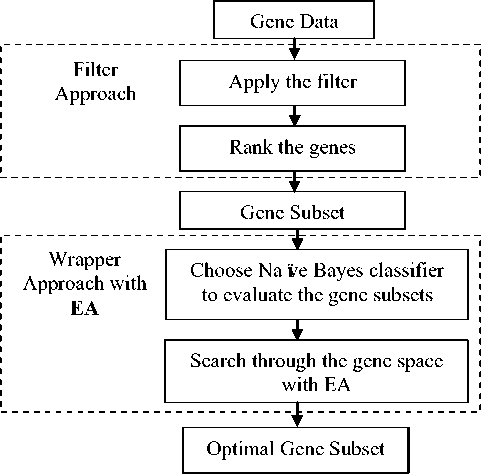
Fig.1. Flow chart of the proposed approach
-
A. Filter Approach
The filter approach in this study is started according to n-1 rule. The top n-1 genes from the original dataset are ranked using a filter with a ranker search strategy. Two filters namely, Information Gain (IG) and Gain Ratio (GR) are initially used on the raw datasets to pick up a gene subset free from redundant and irrelevant genes. IG filter prioritizes the genes based on the concept of entropy where the highest prioritized genes align on the top of the list. On the other hand, GR filter prioritizes the genes based on the gain ratio value of each gene. Unlike wrappers, filters can be applied to a high dimensional dataset regardless its size as it would take a small fraction of time for gene prioritization. Yet, selecting a small informative gene subset is a difficult task only with a filter as filters provide a rank for all the genes by default. Thus, typically filters are used in the preprocessing stage where the dimensionality of the dataset is shrunken. Further, genes are highly correlated with each other emphasizing that the presence of a gene can stimulate the function of another gene which otherwise may be less functional. Thus, selecting a best gene subset is worthwhile.
Typically, filters evaluate each gene alone without considering inter-gene interactions. Nevertheless, wrappers evaluate gene subsets concerning the gene interactions. Therefore, the wrapper approach is introduced followed by the filter approach in this study.
The major drawback of a wrapper is the computational complexity which is increased drastically with the increase of features. The reason is that, the wrappers search through the feature space evaluating all the possible combinations of feature subsets which evolve a huge search space. Due to this issue, the application of a wrapper to an original microarray dataset becomes impractical with the increase of the size of the datasets [2]. As a suggestion, wrappers can be applied with the help of filters in dimensionality reduction.
-
B. Wrapper Approach with Evolutionary Algorithm
The gene subset with highly prioritized n-1 genes enters to the wrapper approach. The wrapper subset evaluator with the Naïve Bayes classifier along with an EA as the search strategy is used in the proposed wrapper approach. The best gene subset which outperforms for the given classifier is resultant as the optimal gene subset at the end.
As a new trend, evolutionary computation techniques are prominently used in microarray analysis [9, 10, 14]. In this study, the evolutionary search which uses the EA is utilized in gene subset selection. It is reflected through the results obtained using the proposed method that the search strategy provides a huge contribution in gene selection. It is ensured through applying discriminant subsets of genes selected from different filters for further analysis using evolutionary search. Though the initial count of genes selected using filters are the same, they are two discriminant set of gene subsets. Nevertheless, wrapper approach is capable of selecting the best informative gene subset which performs well for the given classifier with the contribution of EA. Regardless the size and the elements of the gene subsets selected; they are pretty well in concern to the performance as they provide almost the same performance. More precisely, for instance, in the case of Lymphoma, Information Gain filter with EA (IG-EA) ends up with a gene subset consists of 12 genes whereas Gain Ratio filter with EA (GR-EA) provides a gene subset with 8 genes. Nonetheless, both gene subsets are excellent in terms of performance which is 100%. Further, the default percentage split test option is used to divide the datasets into training and testing sets. The performance is evaluated using two classifiers namely, Naïve Bayes and SVM where both provide the same performance for all the datasets.
-
IV. Results and Discussion
-
A. Experimental Setup
Five microarray datasets namely, Lymphoma, Small Round Blue Cell Tumor (SRBCT), Mixed-Lineage Leukemia (MLL), Leukemia-3 and Colon cancer were used for the evaluation of the proposed approach. The datasets used in this study were obtained from Both binary and multi class problems were considered in this study. Waikato Environment for Knowledge Analysis (WEKA version 3.8.1) the open source machine learning software was used for the implementation. The microarray datasets used for the implementation are illustrated with the number of genes and instances along with a description in Table I.
-
B. Experimental Results
The classification accuracy, number of Correctly Classified Instances (CCI), the total number of instances (between parentheses) and Receiver Operating Characteristic (ROC) value achieved using the proposed method are reported and compared for each dataset. The performance is evaluated using two well-known classifiers; Naïve Bayes and SVM as they outperform in many applications in past research [8, 13, 16, 28]. The number of informative genes selected by wrapper followed by each filter is given in the parenthesis. That is, IG-EA (12) indicates that the number of elements in the gene subset selected by IG-EA for Lymphoma dataset is 12. The classification performance without gene selection and the performance with baseline classifier (i.e. ZeroR classifier) are shown in Table II and Table III respectively.
Table I. The details of cancer microarray datasets used for the evaluation
|
Dataset |
No. of classes |
No. of genes |
No. of samples |
Description |
|
Lymphoma |
3 |
4026 |
66 |
DLBCL: 46, FL: 9 and CLL: 11 |
|
SRBCT |
4 |
2308 |
83 |
EWS: 29, BL: 11, NB: 18 and RMS: 25 |
|
MLL |
3 |
12582 |
72 |
ALL: 24, MLL: 20 and AML: 28 |
|
Leukemia3 |
3 |
7129 |
72 |
B-cell: 38, T-cell: 9 and AML: 25 |
|
Colon |
2 |
2000 |
62 |
Tumor: 40 and Normal: 22 |
Table II. The classification performance of the five cancer datasets without gene selection
|
Dataset |
Naïve Bayes |
SVM |
||||
|
Accuracy (%) |
CCI(Total) |
ROC (%) |
Accuracy (%) |
CCI(Total) |
ROC (%) |
|
|
Lymphoma(4026) |
86.36 |
19(22) |
75.00 |
100.00 |
22(22) |
100.00 |
|
SRBCT(2308) |
96.42 |
27(28) |
97.40 |
100.00 |
28(28) |
100.00 |
|
MLL(12582) |
95.83 |
23(24) |
96.70 |
100.00 |
24(24) |
100.00 |
|
Leukemia3(7129) |
91.66 |
22(24) |
91.00 |
95.83 |
23(24) |
96.70 |
|
Colon(2000) |
52.38 |
11(21) |
56.70 |
80.95 |
17(21) |
88.20 |
Table III. The baseline classification performance of the five cancer datasets
|
Dataset |
ZeroR |
||
|
Accuracy (%) |
CCI(Total) |
ROC (%) |
|
|
Lymphoma(4026) |
69.70 |
46(66) |
50.00 |
|
SRBCT(2308) |
34.93 |
29(83) |
50.00 |
|
MLL(12582) |
38.88 |
28(72) |
50.00 |
|
Leukemia3(7129) |
52.77 |
38(72) |
50.00 |
|
Colon(2000) |
64.51 |
40(62) |
50.00 |
The classification performances with gene selection using both IG-EA and GR-EA methods for Lymphoma, SRBCT, MLL, Leukemia3 and Colon cancer datasets are given in Table IV, Table V, Table VI, Table VII and Table VIII respectively. Note that DLBCL: Diffuse Large B-cell Lymphoma, FL: Follicular Lymphoma, CLL:
Chronic Lymphocytic Leukemia, EWS: Ewing’s Sarcoma, BL: Burkitt’s Lymphoma, NB: Neuroblastoma, RMS: Rhabdomyosarcoma, ALL: Acute Lymphoblastic Leukemia, MLL: Mixed Lineage Leukemia and AML: Acute Myeloid Leukemia in Table I.
Table IV. The classification performance with gene selection for lymphoma dataset
|
Classifier |
IG-EA (12) |
GR-EA (8) |
||||
|
Accuracy (%) |
CCI(Total) |
ROC (%) |
Accuracy (%) |
CCI(Total) |
ROC (%) |
|
|
Naïve Bayes |
100.00 |
22(22) |
100.00 |
100.00 |
22(22) |
100.00 |
|
SVM |
100.00 |
22(22) |
100.00 |
100.00 |
22(22) |
100.00 |
Table V. The classification performance with gene selection for srbct dataset
|
Classifier |
IG-EA (21) |
GR-EA (17) |
||||
|
Accuracy (%) |
CCI(Total) |
ROC (%) |
Accuracy (%) |
CCI(Total) |
ROC (%) |
|
|
Naïve Bayes |
100.00 |
28(28) |
100.00 |
100.00 |
28(28) |
100.00 |
|
SVM |
100.00 |
28(28) |
100.00 |
100.00 |
28(28) |
100.00 |
Table VI. The classification performance with gene selection for mll dataset
|
IG-EA (15) |
GR-EA (16) |
|||||
|
Accuracy (%) |
CCI(Total) |
ROC (%) |
Accuracy (%) |
CCI(Total) |
ROC (%) |
|
|
Naïve Bayes |
100.00 |
24(24) |
100.00 |
100.00 |
24(24) |
100.00 |
|
SVM |
100.00 |
24(24) |
100.00 |
100.00 |
24(24) |
100.00 |
Table VII. The classification performance with gene selection for leukemia3 dataset
|
Classifier |
IG-EA (13) |
GR-EA (17) |
||||
|
Accuracy (%) |
CCI(Total) |
ROC (%) |
Accuracy (%) |
CCI(Total) |
ROC (%) |
|
|
Naïve Bayes |
95.83 |
23(24) |
100.00 |
95.83 |
23(24) |
99.30 |
|
SVM |
95.83 |
23(24) |
95.90 |
95.83 |
23(24) |
97.20 |
Table VIII. The classification performance with gene selection for colon cancer dataset
|
Classifier |
IG-EA (16) |
GR-EA (17) |
||||
|
Accuracy (%) |
CCI(Total) |
ROC (%) |
Accuracy (%) |
CCI(Total) |
ROC (%) |
|
|
Naïve Bayes |
90.47 |
19(21) |
97.10 |
90.47 |
19(21) |
96.60 |
|
SVM |
90.47 |
19(21) |
94.10 |
90.47 |
19(21) |
94.10 |
Table IV, V, VI, VII and VIII show the classification performance with gene selection for Lymphoma, SRBCT, MLL, Leukemia and Colon cancer respectively. From the observation of Table IV, Table V and Table VI, the results obtained indicate that the proposed approach has given the highest performance for the relevant datasets. Though, Leukemia3 and Colon cancer classification performances are a bit lower compared to that of other three datasets, they are still capable to be classified with only one and two misclassifications respectively. With comparison to the study reported in [4, 12], the proposed study has obtained little bit higher accuracy which is 90.47% for colon cancer dataset whereas it is 90.32% with 3 genes in former and 90.09% with 30 genes in later.
Further, in the classification of colon cancer, a sparse representation based method is proposed in [3] which provide 91.94% accuracy; nevertheless with a very huge gene subset. Moreover, the accuracies for colon cancer dataset and SRBCT are greater than [9, 29]. At the same time the performance of SRBCT is greater than that of reported in [30]. The proposed approach has obtained the perfect classification performance for SRBCT dataset similar to the algorithm proposed by [28], even though [28] provides somewhat higher accuracy for Colon cancer dataset than what is gained in this study. Further, in the comparison of performance for Leukemia3 dataset, the proposed method in [9] achieved 96.64% accuracy whereas nearly a similar accuracy 95.83% is obtained in this study. A small number of gene subset is used in [31] in the classification of SRBCT and MLL which achieved the same accuracy as the proposed method.
Despite of that the number of genes in each gene subsets slightly vary in their count, both subsets selected by IG-EA and GR-EA provide the same classification accuracies indicating that they are best in terms of performance. Moreover, both classifiers gave the same performance for all the datasets though they use discriminant algorithm for evaluations. This achievement further validates the mentioned study and the predictability of the genes selected. The total time spent for both IG-EA and GR-EA are shown in Table IX. The results indicate that the approach is appropriate even for very huge datasets as it is in few seconds.
Table IX. The average runtime (in second) for the proposed approach
|
Dataset |
IG-EA (s) |
GR-EA (s) |
|
Lymphoma |
7 |
3 |
|
SRBCT |
9 |
8 |
|
MLL |
4 |
4 |
|
Leukemia3 |
5 |
5 |
|
Colon |
4 |
2 |
-
V. Conclusion
In this study, a new hybrid method with wrappers and filters is proposed. The proposed approach points out the effect of using EA in feature subset selection. Two different filters along with a wrapper namely, IG-EA and GR-EA are used to evaluate the performance of classification on five microarray datasets. Gene subset selected through a wrapper approach highly dominated by the classification algorithm used to search through the feature space and the classifier used to evaluate each gene subset. Thus, a comparatively best approach is supposed in this paper for gene selection. The paper suggests a computationally less complex search strategy with an evolutionary search and Naïve Bayes classifier. Both binary classification and multi class classification problems are concerned to be assessed. To overcome the issues with huge datasets, the filter base preprocessing is carried out initially before using the wrapper. Once the size of the data set is reduced through preprocessing the application of a wrapper is made easier. Thus, regardless the size of the dataset, the proposed approach can be applied even to a huge microarray dataset without penalization of time. The evaluation results showed in Table II-IX indicate that the proposed approach is reliable and efficient in terms of classification performance as three out of five datasets achieve 100% accuracy with a small number of genes and less computational expense.
References The effect of evolutionary algorithm in gene subset selection for cancer classification
- I. Guyon, J. Weston, S. Barnhill, and V. Vapnik, “Gene selection for cancer classification using support vector machines,” Machine learning, vol. 46, no. 1, pp. 389–422, 2002.
- C. Kim, H. Li, S.-Y. Shin, and K.-B. Hwang, “An efficient and effective wrapper based on paired t-test for learning naive bayes classifiers from large-scale domains,” Procedia Computer Science, vol. 23, pp. 102–112, 2013.
- B. Gan, C.-H. Zheng, J. Zhang, and H.-Q. Wang, “Sparse representation for tumor classification based on feature extraction using latent low-rank representation,” BioMed research international, vol. 2014, 2014.
- R. Aziz, C. Verma, and N. Srivastava, “A fuzzy based feature selection from independent component subspace for machine learning classification of microarray data,” Genomics data, vol. 8, pp. 4–15, 2016.
- T. Golub, D. Slonim, P. Tamayo, C. Huard, M. Gaasenbeek, J.H.H.C. Mesirov, M. Loh, J. Downing, M. Caligiuri, C. Bloomfield, and E. Lander, “Molecular classification of cancer: class discovery and class prediction by gene expression monitoring”. Science, 286 (5439), pp. 531–537, Oct. 1999.
- T.S. Furey, N. Cristianini, N. Duffy, D.W. Bednarski, M. Schummer, D. Haussler, “Support vector machine classification and validation of cancer tissue samples using microarray expression data”. Bioinformatics, vol. 16, pp. 906–914, Oct. 2000.
- I. Guyon, J. Weston, S. Barnhill, and V. Vapnik, “Gene selection for cancer classification using support vector machines,” Machine learning, vol. 46, no. 1, pp. 389–422, 2002.
- Y. Wang, I. V. Tetko, M. A. Hall and E. Frank, “Gene selection from microarray data for cancer classification: A machine learning approach”, Comput Biol Chem, vol. 29, pp. 37-46, Feb. 2005.
- Zexuan Zhu, Y. S. Ong and M. Dash, “Markov Blanket-Embedded Genetic Algorithm for Gene Selection”, Pattern Recognition, Vol. 49, No. 11, 3236-3248, 2007.
- M. Sun, L. Xiong, H. Sun, and D. Jiang, “A ga-based feature selection for high-dimensional data clustering,” in Genetic and Evolutionary Computing, 2009. WGEC’09. 3rd International Conference on. IEEE, 2009, pp. 769–772.
- S. H. Bouazza, K. Auhmani, A. Zeroual, and N. Hamdi, “Selecting significant marker genes from microarray data by filter approach for cancer diagnosis,” Procedia Computer Science, vol. 127, pp. 300–309, 2018.
- L. Gao, M. Ye, X. Lu, and D. Huang, “Hybrid method based on information gain and support vector machine for gene selection in cancer classification,” Genomics, proteomics & bioinformatics, vol. 15, no. 6, pp. 389–395, 2017.
- M. N. F. Fajila and R. D. Nawarathna, “New feature selection method for high dimensional gene data,” in Symposium on Statistical & Computational Modelling With Applications, Department of Statistics & Computer Science, University of Kelaniya, Sri Lanka, Nov. 2016, pp. 67–70.
- C. De Stefano, F. Fontanella, and A. S. di Freca, “Feature selection in high dimensional data by a filter-based genetic algorithm,” in European Conference on the Applications of Evolutionary Computation. Springer, 2017, pp. 506–521.
- C.-S. Yang, L.-Y. Chuang, C.-H. Ke, and C.-H. Yang, “A hybrid feature selection method for microarray classification.” IAENG International Journal of Computer Science, vol. 35, no. 3, 2008.
- K. DAS and D. MISHRA, “Hybridized univariate and multivariate filter based approaches for gene selection,” International Journal of Pharma and Bio Sciences, vol. 7, 2016.
- P. A. Mundra and J. C. Rajapakse, “Svm-rfe with mrmr filter for gene selection,” IEEE transactions on nanobioscience, vol. 9, no. 1, pp. 31–37, 2010.
- C. A. Kumar, M. Sooraj, and S. Ramakrishnan, “A comparative performance evaluation of supervised feature selection algorithms on microarray datasets,” Procedia Computer Science, vol. 115, pp. 209–217, 2017.
- E. Alba, J. Garcia-Nieto, L. Jourdan, and E.-G. Talbi, “Gene selection in cancer classification using pso/svm and ga/svm hybrid algorithms,” in Evolutionary Computation, 2007. CEC 2007. IEEE Congress on. IEEE, 2007, pp. 284–290.
- H. Motieghader, A. Najafi, B. Sadeghi, and A. Masoudi-Nejad, “A hybrid gene selection algorithm for microarray cancer classification using genetic algorithm and learning automata,” Informatics in Medicine Unlocked, vol. 9, pp. 246–254, 2017.
- F. Jimenez, G. Sanchez, J. M. Garcia, G. Sciavicco, and L. Miralles, “Multi-objective evolutionary feature selection for online sales forecasting,” Neurocomputing, vol. 234, pp. 75–92, 2017.
- W. Xiong and C. Wang, “A hybrid improved ant colony optimization and random forests feature selection method for microarray data,” in Proc. International Conference on Networked Computing and Advanced Information Management, pp. 559–563, 2009.
- B. Sahu and D. Mishra, “A novel feature selection algorithm using particle swarm optimization for cancer microarray data,” Procedia Engineering, vol. 38, pp. 27–31, 2012.
- H. M. Alshamlan, “Co-abc: Correlation artificial bee colony algorithm for biomarker gene discovery using gene expression profile,” Saudi Journal of Biological Sciences, 2018.
- Alshamlan, H.M., Badr, G.H., Alohali, Y.A., 2016. Abc-svm: artificial bee colony and svm method for microarray gene selection and multi class cancer classification. Int. J. Mach. Learn. Comput. 6 (3), 184.
- H. Alshamlan, G. Badr, and Y. Alohali, “mrmr-abc: a hybrid gene selection algorithm for cancer classification using microarray gene expression profiling,” BioMed research international, vol. 2015, 2015.
- M. Toghraee, H. Parvin, and F. Rad, “The impact of feature selection on meta-heuristic algorithms to data mining methods,” International Journal of Modern Education and Computer Science(IJMECS), vol. 8, no. 10, p. 33, 2016. DOI: 10.5815/ijmecs.2016.10.05
- H. M. Alshamaln, “Dqb: a novel dynamic quantitive classification model using artificial bee colony algorithm with application on gene expression profiles,” Saudi Journal of Biological Sciences, 2018.
- H. Vural and A. Subas¸ı, “Data-mining techniques to classify microarray gene expression data using gene selection by svd and information gain,” Model Artificial Intel, vol. 6, pp. 171–182, 2015.
- M. Mramor, G. Leban, J. Demˇsar, and B. Zupan, “Visualization-based cancer microarray data classification analysis,” Bioinformatics, vol. 23, no. 16, pp. 2147–2154, 2007.
- A. Sharma, S. Imoto, and S. Miyano, “A top-r feature selection algorithm for microarray gene expression data,” IEEE/ACM Transactions on Computational Biology and Bioinformatics (TCBB), vol. 9, no. 3, pp. 754–764, 2012.
- T. Li, C. Zhang, and M. Ogihara, “A comparative study of feature selection and multiclass classification methods for tissue classification based on gene expression,” Bioinformatics, vol. 20, no. 15, pp. 2429–2437, 2004.
- M. Panda, “Elephant search optimization combined with deep neural network for microarray data analysis,” Journal of King Saud University-Computer and Information Sciences, 2017.
- S. Roy and S. S. Chaudhuri, “Cuckoo search algorithm using levy flight: a review,” ´ International Journal of Modern Education and Computer Science, vol. 5, no. 12, p. 10, 2013.
- S. Roy and S. S. Chaudhuri, “Bio-inspired ant algorithms: A review,” International Journal of Modern Education and Computer Science, vol. 5, no. 4, p. 25, 2013.
- Sergii Babichev, Jiří Škvor, Jiří Fišer, Volodymyr Lytvynenko, "Technology of Gene Expression Profiles Filtering Based on Wavelet Analysis", International Journal of Intelligent Systems and Applications(IJISA), Vol.10, No.4, pp.1-7, 2018. DOI: 10.5815/ijisa.2018.04.01
