The types of silicon–oxygen bonds from the perspective of descriptors based on one-electron potentials

Author: Klyuev I.V., Bartashevich E.V.
Journal: Вестник Южно-Уральского государственного университета. Серия: Химия @vestnik-susu-chemistry
Section: Физическая химия
Article in issue: 1 т.18, 2026.
Free access
The properties of multicomponent compounds are determined by the types and characteris-tics of chemical bonds, enabling the study of the nature of interatomic interactions in different phases of matter: from gases to solids, including a crystal phase. Copolymers based on polydimethylsiloxanes are widely used because of their chemical resistance, relative inertness, and ability to spontaneously restore their initial structure through a self-healing process after defect formation. This is experienced due to the so-called “siloxane equilibrium” phenomenon, in which the O–Si bond in a polysiloxane chain can dissociate to form the anion that can attack another chain leading to crosslinking and dynamic chain exchange. Since the ability for self-healing mainly depends on the formation of both covalent and noncovalent bonds; it is essential to develop quantitative criteria for the identification of the bond types in multiscale models. There-fore, the main goal of this study is to analyze the electronic descriptors for classifying different types of sili-con-oxygen bonds. For this purpose, we have modeled and analyzed various molecular and crystal structure models containing O–Si/O...Si bonds of different types. These bonds were categorized using electronic de-scriptors previously developed within the framework of the orbital-free density functional theory (OF-DFT) and based on one-electron potentials. One of these descriptors involves the comparison of the gaps between the positions of 1D extremes in electron density, electrostatic potential and total static potential functions along the interatomic line, which makes it possible to identify covalent bonds, coordination bonds and weak noncovalent interactions (or the tetrel bonds, TtB). The aim of our research is to determine the applicability of the electronic criterion and evaluate descriptors which enables identification of strong covalent (O–Si) or coordination bonds and weak (O...Si) noncovalent interactions in various systems containing silicon–oxygen interactions. We have started by analyzing the absolute difference between the positions of ex-tremes in electronic functions, which have allowed the identification of bond types. Next, we have analyzed the features of identical compounds in different phase states that affect the electronic structure and descrip-tor values, and also determined the impact of the formal charge on the values of electronic descriptors.
Orbital-free DFT, tetrel bond, silicon, electronic criterion, two-factor rule
Short address: https://sciup.org/147253377
IDR: 147253377 | UDC: 544.144 | DOI: 10.14529/chem260111
Типы связей кремний–кислород с позиции дескрипторов на основе одноэлектронных потенциалов
Свойства многокомпонентных соединений во многом определяются типами и характеристиками химических связей, которые, в свою очередь, позволяют изучить природу межатомных взаимодействий в разных фазовых состояниях вещества: от газовой фазы до кристалла или твердого тела. Сополимеры на основе полидиметилсилоксанов широко применяются благодаря их химической стойкости, относительной инертности, а также способности самопроизвольно восстанавливать свою структуру после появления дефектов. Известен эффект так называемого «силоксанового равновесия», при котором связь O–Si в полисилоксановой цепи может диссоциировать с образованием аниона, который может атаковать другую цепь, позволяя формироваться сшивкам и протекать динамическому обмену. Поскольку способность к самовосстановлению во многом зависит от формирования как ковалентных, так и нековалентных связей, полезным представляется иметь количественные критерии, которые позволят идентифицировать типы связей в многомасштабных моделях. Поэтому разработка электронных дескрипторов, нацеленных на установление типов связей кремний – кислород выступила основной целью данной работы. Мы смоделировали и проанализировали различные молекулярные и кристаллические структуры, содержащие связи O–Si/O...Si разных типов. За основу их категоризации были взяты ранее разработанные в рамках неорбитальной теории функционала плотности (OFDFT) электронные дескрипторы, вычисляемые на основе одноэлектронных потенциалов. Один из них включает сравнение длин отрезков между позициями экстремумов функций электронной плотности, электростатического потенциала и полного статического потенциала вдоль межатомной линии, что позволяет идентифицировать сильные ковалентные, координационные и слабые нековалентные связи. Задачей работы явилась проверка применимости электронного критерия и оценка дескрипторов, которые позволяют идентифицировать сильные ковалентные (O–Si), координационные и слабые (O...Si) нековалентные связи в самых разнообразных системах, содержащих кремнийкислородные взаимодействия. Для этого мы прибегли к анализу абсолютной разницы между позициями экстремумов электронных функций, которая позволяет идентифицировать типы связей. Проанализированы особенности, влияющие на электронное строение и величины дескрипторов для одинаковых соединений, находящихся в разных фазовых состояниях, а также определено влияние заряда системы на величины электронных дескрипторов.
Text of the scientific article The types of silicon–oxygen bonds from the perspective of descriptors based on one-electron potentials
Chemical bonds determine the structure of a molecule, which in turn influences their physicochemical properties. A chemical bond is an interaction between two atoms. The description of its electronic structure can be considered from two perspectives: either valence bond theory [1] or molecular orbital theory [2]. Depending on whether covalent bonding or noncovalent interaction takes place, the electronic properties of the bond and the interaction mechanism between atoms differ.
Nowadays, the classification of noncovalent interactions is a hot topic. One such suggests categorizing them by the group which an element belongs to. Thus, IUPAC provides definitions for halogen [3], chalcogen [4] and pnictogen bonds [5]. The development of the nomenclature for tetrel bonds is a central focus in the field of theoretical chemistry [6, 7]. The taxonomy of these kinds of interactions involves the identification of the group of an electrophilic atom, which gives the name to the chemical bond, but also geometric features that allows the classification of the contact between atoms providing an electrophilic site by type I and type II [8]. The tetrel bond is a type of interaction that occurs between an electrophilic site, provided by the group 14 atom, and a nucleophilic fragment [9]. The atoms that have a predisposition to form the tetrel bonds have the deepest σ-hole [10].
The increasing interest in the bonds between silicon and oxygen atoms can be driven by the properties exhibited by polysiloxane-based polymers. In particular, copolymers of polydimethylsiloxane and metal complexes exhibit unique self-healing properties which can be modified by copolymerization with other compounds [11, 12]. However, the exact mechanism of self-healing and the processes that affect it remain to be studied [12–14]. It is known that the dynamics of O–Si bonds formation and breaking plays a significant role observed in a phenomenon of the so-called “siloxane equilibrium”. The chemical reaction mechanism is illustrated in Fig. 1 [15–17]. It is based on the reversible reorganization of broken polymer chains along the O–Si bond, which is affected by various catalysts, such as hydroxides, water, carbenes, cations and initiator residues [18,19].

Fig. 1. Dynamic rearrangement of chain fragments via breaking and formation of O–Si bonds in the “siloxane equilibrium” polymers, where R = –(OSi(CH 3 ) 2 ) n –
The mechanism of the “siloxane equilibrium” involves breaking a O–Si bond within the polysiloxane chain and formation of silanolate anion O–SiR3–, which has the ability for regrouping with other segments of the chain through nucleophilic attack on silicon atoms in other siloxane chains [20]. In our study, the O–Si bond was chosen, firstly, to gain a deeper understanding of the nature of tetrel bonds – bonds formed by atoms from the carbon group that act as electrophiles, influencing the nucleophilic addition-cleavage reactions in copolymers based on polydimethylsiloxane. And secondly, there are numerous materials containing organosilicon compounds which exhibit unique properties in catalytic and synthetic processes. Additionally, polysiloxane-based polymers have flexible modification capabilities that can possess a wide variety of physicochemical properties in materials, including thermal stability and desired permittivity ranges.
The properties of chemical bonds and their classification into different types can be investigated using orbital-free density functional theory (OF-DFT) [21, 22], where various approximate kinetic energy density functionals expressed in terms of the electron density are used and Kohn-Sham orbitals can be viewed as means to perform self-consistent density calculations in OF-DFT functionals, thereby reducing computational costs [23, 24].
Current concepts and methods developed at the intersection of OF-DFT and QTAIM have proven fruitful for understanding the nature and types of chemical bonds. These methods are particularly useful for identifying noncovalent interactions, such as those involving tetrel, pnictogen, chalcogen and halogen atoms. The categorization of the types of chemical bonds can be performed using the electronic criterion [25], which is connected to analysis of dispositions of electron density and electrostatic potential minima and total static potential maximum along the interatomic lines. Then the width of gaps between the extreme values of electronic functions [26] are measured and their values are compared. If we denote the gap between υ st (r) max and υ els (r) min positions as ∆ st-els , and the gap between υ st (r) max and ρ (r) min positions as ∆ ρ -st , then for short, strong bonds ∆ st-els is smaller than ∆ ρ -st , and for typical noncovalent interactions, vice versa , ∆st-els is larger than ∆ ρ -st. The two-factor rule itself allows for the distinction between noncovalent interactions, covalent bonds and coordination bonds. Thus, the dependence of the absolute difference, |∆ st-els – ∆ ρ -st |, on the interacting atoms distance reveals three zones on the scatterplot. At the boundaries between these zones bond properties change, indicating a change in bond type.
Such analysis has been performed also for tetrel bonds. For example, in these studies [27, 28] the electronic criterion is used for identification of the bonds with tetrel atoms in tetrahedral molecular complexes and in crystals while varying the halogen nucleophilic fragment X and the substituents along the bond line Y4Tt...X.
The goal of our theoretical study is to categorize O–Si/O...Si chemical bonds using the structural models of molecular complexes and crystals, and to establish criteria, based on QTAIM and OF-DFT approaches, that appropriate for identification of the bond type (covalent, coordination or noncovalent) for Si–O interaction in complex multicomponent systems. For these purposes, we set the following tasks: 1) to create a diverse set of compounds containing different types of O–Si/O...Si bonds in the composition of gas-phase molecular complexes and crystals; 2) to test the applicability of the two-factor rule and the electron criterion that was developed using the examples of N–Si/N...Si, Hal–Si/Hal...Si (Наl = F, Cl, Br) [26, 28] on the O–Si/O...Si bonds; 3) to determine factors that influence the absolute difference value for the bonds of the same length; 4) to compare Si–O interactions in the same compound in different phases: in gas-phase complexes and in crystals.
The objects for modeling included in the set are: tetrahedral Y–SiX 3 complexes (X,Y = OH, OCH 3 , H, F, Cl, Br, propyl, phenyl) within the gas-phase approximation, where an oxygen-containing fragment (OR) acting as a nucleophile was positioned along the interatomic line Y–Si, where on the opposite side of Si was a σ-hole – a region with increased values of electrostatic potential (OR = OH–, OCH 3 –, HSO 3 –, HSO4–, H2O, CO, CO2, CH3OH, epoxy); silatranes within the gas-phase approximation and in crystal forms with various substituents at the four-coordinated silicon atom, where one or two H2O molecules were placed near the O–Si bonds in the gas-phase approximation, leading to the hydrolysis reaction with subsequent partial opening of the silatrane framework [29]; spirosilanes and its derivatives [30]; monochelate complexes obtained by the transsilylation of silylated pyridones [31] and complexes of pentacoordinated oligosilanes with N -methyl- N -trimethylsilylacetamide [32]; triflate complex with terpyridine-stabilized monophenylsilyl [33, 34]; 2D-periodic systems of graphene-like silicon carbide monolayers with a vacancy-type defect, interacting with 1-methoxysilatrane [35, 36]. All compounds, except for tetrahedral complexes, were taken from open databases. Representatives of the set studied in our research are shown in Fig. 2.
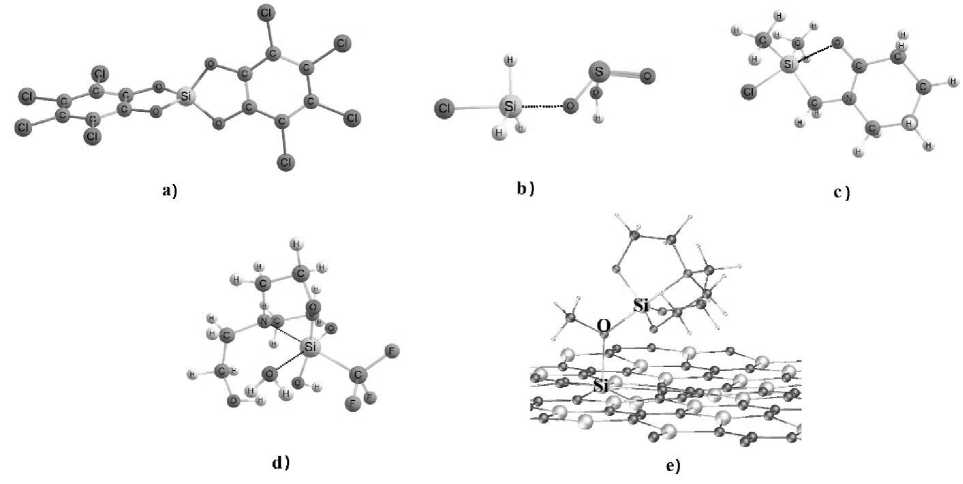
Fig. 2. Typical representatives of compounds in our set: a – perchloro-substituted spirosilane [37];
b – tetrahedral complex of monochlorosilane [38] with bisulfite; c – complexes derived from silylated pyridones [39];
d – 1-(trifluoromethyl)silatrane [40] after interacting with two H 2 O molecules;
e – 1-methoxysilatrane [41] on the silicon carbide surface with a vacancy-type defect
The equilibrium state structures in the gas-phase approximation for molecular complexes have been obtained by performing geometry optimization in the FireFly 8.2.0 program [42] using the PBE0 hybrid functional [43] with Jorge-DZP [44] basis set from the Basis Set Exchange site [45]. For systems containing bromine atoms the Douglas-Kroll-Hess approach has been employed [46]. Gradient convergence was set to 0.9 x 10–6. The optimized structures have been tested for the absence of imaginary IR frequencies.
The optimization of crystals and complexes with silicon carbide surfaces has been performed using only atomic coordinates in the CRYSTAL17 software package [47] with the pob-DZVP-rev2 [48] basis set using the PBE0 functional with the following convergence parameters: energy change in the self-consistent field calculations – 10–6 a.u., displacement of atomic coordinates – 10–3 a.u., force gradient – 10–3 a.u.
A quantum topological analysis of the electron density has been carried out and electronic properties such as the electrostatic potential, υ els (r) and the potential acting on the electron in a molecule (or the total static potential), υ st (r) [49] have been calculated for all compounds including gas-phase complexes, crystals and 2D-periodic surfaces. In order to establish interactions between Si and O atoms we have calculated bond paths and bond critical points (BCP) (3; –1) [50]. For molecular complexes in the gasphase approximation we have used electronic characteristics such as electron density ρ (r), electrostatic potential υ els (r) and total static potential υ st (r) [51], that we have calculated in the Multiwfn 3.8 [52] program, to analyze chemical bonding within the framework of the quantum theory of atoms in molecules (QTAIM) [53]. For compounds in crystal form we have used the WinXPRO [54] program with the Barth-Hedin exchange potential [55] and the Chachiyo-Karasiev correlation potential [56, 57] calculated from electron density.
To determine the applicability of the two-factor rule for O–Si/O...Si bonds the values of the absolute difference between ∆st-els и ∆ ρ -st gaps in one-dimensional distribution of functions such as electron density, total static and electrostatic potentials were estimated along the bond lines. We have confirmed that according to the electronic criterion [26] for weak O...Si bonds ∆ st-els > ∆ ρ -st , and for strong O–Si bonds, it is, vice versa , ∆st-els< ∆ ρ -st, where xst, xels и x ρ are the extreme positions of corresponding functions along the line connecting two atoms.
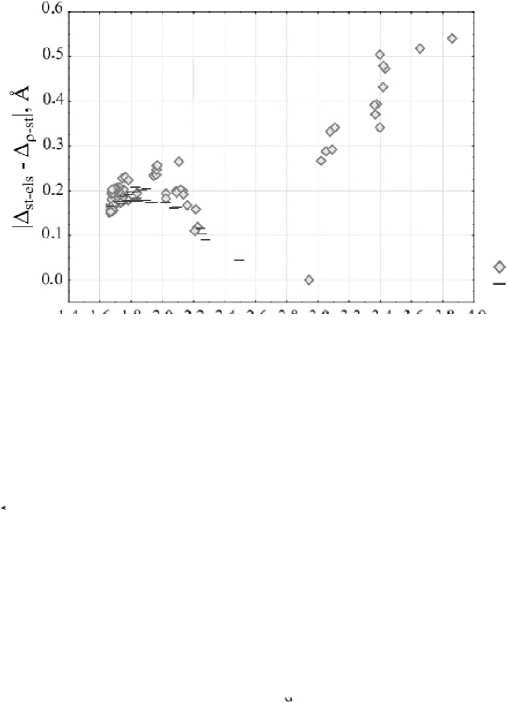
For all O–Si/O...Si in our set we present the scatterplot in Fig. 3 in |∆ st-els – ∆ ρ -st | vs d coordinates, where d denotes the distance between O и Si. As can be seen, the datapoints on the graph in these coordinates form a particular line that has 2 extremes, which were used by the authors [58] as possible range boundaries for different bond types, transitioning from covalent bonding to noncovalent interaction.
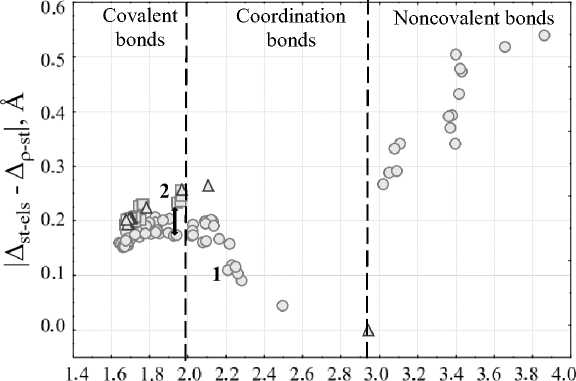
О In molecular complexes A In 2D-periodic surfaces □ In crystals
Fig. 3. Scatterplot of O–Si/O...Si bonds with the absolute difference value against the bond lengths in compounds, modeled in the gas-phase approximation, as crystals or as 2D-periodic surfaces, with the illustration of three ranges, characterizing the type of the interatomic interaction. The number 1 is N -methyl- N -
(dimethylchlorosylilmethyl)acetamide [59], modeled in the gas-phase approximation; the number 2 – modeled as a crystal
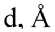
The first range includes O–Si bonds with the shortest lengths, and the absolute difference |∆st-els – ∆ ρ -st| criterion increases with increasing the bond length for them. Such bonds are covalent and for our set their length range is from 1.64 Å to 1.97 Å with absolute difference value changing from 0.15 Å to 0.26 Å. In the next range of distances d values of |∆st-els – ∆ ρ -st| decrease as bond lengths increase. It is assumed, that a donor-acceptor mechanism is involved in this group of compounds in the O–Si bond formation, and the bonds are donor-acceptor or coordination, and their lengths range is from 2.02 Å to 2.94 Å with |∆ st-els – ∆ ρ -st | values decreasing from 0.27 Å to 0 Å. The value of the absolute difference being 0 can be explained that in this case, where ∆ st-els = ∆ ρ -st , according to the electronic criterion the O–Si bond in this compound has an intermediate bond strength value between covalent bonds and noncovalent interactions, that is, it is a typical example of the interaction by donor-acceptor mechanism. And, lastly, in the third range at large interatomic distances where there is no significant electron distribution between O и Si atoms noncovalent interactions are involved. The values of |∆ st-els – ∆ ρ -st | for them increase with O...Si bond lengths increase. Most of these could be attributed to tetrel bonds, if we consider the electrostatic nature of the interaction in which the Si atom form a σ -hole, interacting with the lone electron pair of the O atom.
If we examine in more detail the range with covalent bonds in Fig. 3, we can identify certain trends for O–Si bonds of compounds in crystals, in molecular complexes in the gas-phase approximation and in molecular complexes interacting with surfaces, for which geometry optimization was processed taking into account periodic boundary conditions. That is, for close values of d there is a difference of |∆st-els – ∆ ρ -st | for them, and this trend holds for the entire range of covalent bonds. Let's compare for example the molecular complex of chlorosilane with bisulfite and the crystal structure of 5-chloro-2,3-dimethyl-5,5- bis (trimethylsylil)-1-oxa-3-aza-5-silacyclopentene [32], for which a close O–Si bond length of 1.94 Å is observed. For crystal structure |∆st-els – ∆ ρ -st| = 0.23 Å, and for the molecular complex |∆st-els – ∆ ρ -st| = 0.18 Å. It is possible that, in addition to the structural characteristics of each compound, the effect of the crystal environment also plays a role, which typically results in shorter bonds with stronger electronic interaction between bonded atoms. Furthermore, we have noticed that the same compound can exhibit significantly different bond characteristics depending on its aggregate state. In Fig. 4 there are distributions of electron density and static and electrostatic potentials values for intramolecular O–Si bond in N -methyl- N -(dimethylchlorosylilmethyl)acetamide, modeled as an isolated molecule in the gas-phase approximation and as a crystal with periodic boundary condition. Due to the specific arrangement of molecules within a crystal lattice, the O–Si bond has a shorter bond length (1.96 Å) in this case, than in the isolated molecule (2.23 Å) despite the use of identical DFT functionals and basis sets. Due to the shift in the position of the static potential maximum towards the minimum of the electron density position for the bond in в gas phase approximation there is a change in ∆st-els and ∆ ρ -st values (0.14 Å and 0.26 Å respectively for crystal and 0.06 and 0.30 Å respectively for molecular complex), that leads to the change in |∆ st-els – ∆ ρ -st | from 0.12 Å to 0.24 Å. And, according to the two-factor rule, this bond should be covalent in the crystal and coordination in the isolated molecule, as we see this bond in different ranges in Fig. 3. Similar findings were previously reported for silatranes and germatranes in the article [60].
Fig. 4. The structure and the extreme positions of electron density, electrostatic potential and total static potential functions along the O–Si bond line in N -methyl- N -(dimethylchlrorosylilmethyl)acetamide [59]: on the left – in the gas-phase approximation, on the right – in the crystal. The solid line is an electron density; the dotted line is an electrostatic potential function; the solid line with dots is a total potential function. The vertical lines indicate the extreme positions of the corresponding functions
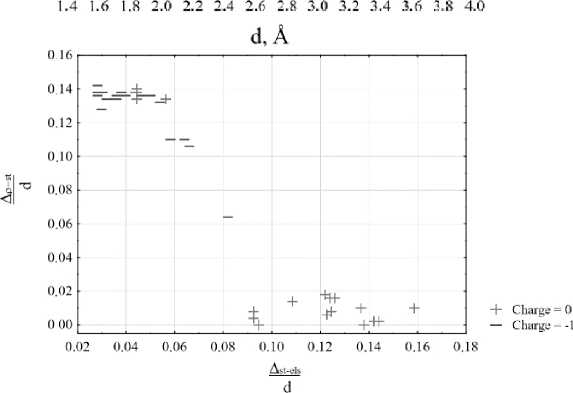
Another interesting observation can be made if show the formal charges of the complexes on the scatterplot. The set includes both neutral and negatively charged molecular complexes. Fig. 5a illustrates that negatively charged complexes have lower |∆ st-els – ∆ ρ -st | values. This is due to the fact that in negatively charged systems containing one extra electron, there is a displacement in the electron density minimum and potentials extremes positions, but for different gaps ∆ st-els and ∆ ρ -st the extent if this displacement varies ∆st-els gap for the systems with negative formal charge is generally shorter than for the neutral systems and ∆ ρ -st gap is wider, but ∆ ρ -st value changes more than ∆ st-els value. In order to facilitate the comparison, we proceeded to reduced values by dividing the ∆ st-els and ∆ ρ -st values by the corresponding bond lengths values. From Fig. 5b it can be observed, that for the most of negatively charged systems there is a displacement towards lower values of ∆ st-els ( ∼ 8%) and higher values of ∆ ρ -st ( ∼ 12%). And due to the different changes in the values of these gaps |∆ st-els – ∆ ρ -st | varies unevenly and has a lower value. However, there are some exceptions for the tetrafluorosilane and trimethoxyhydroxysilane complexes, where the behavior of ∆ st-els and ∆ ρ -st gaps values can be influenced by conformations where the region for the O...Si bond path is affected by nearby lone electron pairs from F or O substituents, which can be confirmed by significantly overestimated electron density minima values ( ∼ 7 times greater) electrostatic potential minima values ( ∼ 10 times greater) and underestimated total static potential maxima values ( ∼ 2 times greater) compared to the other neutral complexes.
a)
Charge = О
Charge — - I
b)
In the context of the orbital-free DFT, we conducted an analysis of electronic potentials in a one-electron approximation approach, which demonstrated the capacity to categorize the O–Si/O...Si bond types using the extreme positions of electron density, electrostatic and total static potentials in 1D distributions along interatomic lines.
This analysis was performed using the two-factor rule's descriptors based on the absolute difference between the gaps that are defined by extremes of electrostatic and total static potentials, differently distant from the boundary of atomic basins. This approach allowed us categorizing the interatomic interactions between silicon and oxygen atoms into one of three groups: covalent bonds, coordination bonds and noncovalent interactions (tetrel bonds). Furthermore, we established that the values of the absolute difference, |∆ st-els – ∆ ρ -st |, can vary depending on a system charge and on a phase of compound, whether it is a crystal or an isolated molecule in a gas-phase. Specifically, in negatively charged systems, there is a reduction in the absolute difference value of the O–Si bond. Additionally, we managed to determine that in crystals a O–Si bond has a larger absolute difference value than a bond with the same length of a compound in the gas-phase approximation. Finally, we found that the bonds within identical structures, but in different phases, can belong to different types.
