Влияние базисной терапии ревматоидного артрита на активность апоптоза периферических лимфоцитов

Автор: Рябков Вадим Александрович, Везикова Наталья Николаевна, Барышева Ольга Юрьевна, Волкова Татьяна Олеговна
Журнал: Ученые записки Петрозаводского государственного университета @uchzap-petrsu
Рубрика: Медицинские науки
Статья в выпуске: 7 (101), 2009 года.
Бесплатный доступ
Ревматоидный артрит, апоптоз, лимфоциты, каспазы, базисная терапия, метотрексат
Короткий адрес: https://sciup.org/14749608
IDR: 14749608
Текст статьи Влияние базисной терапии ревматоидного артрита на активность апоптоза периферических лимфоцитов
Ревматоидный артрит – хроническое воспалительное заболевание, которое характеризуется преимущественным поражением суставов в виде синовиальной гиперплазии с местной инфильтрацией различными клетками воспаления, что ведет к необратимой деструкции хряща, костной ткани и нередко сочетается с системным воспалительным поражением внутренних органов [6], [13], [17], [23], [36].
РА является самой частой аутоиммунной патологией человека и одним из наиболее распространенных воспалительных заболеваний суста-
вов. РА поражает до 0,5–1,0 % населения, отличается неблагоприятным прогрессирующим течением – через 10–15 лет от начала болезни трудоспособность теряют примерно 90 % пациентов [11], [15], [17].
Этиология РА достоверно неизвестна. Сложность и многофакторность патогенеза заболевания побуждают к поиску новых методов исследования, позволяющих оптимизировать проводимое лечение, которое в настоящее время у части больных не оказывает достаточного эффекта. В основе патологического процесса при РА лежит активация CD4+ Т-клеток неизвестным антигеном у генетически предрасположенных лиц с увеличением размеров клона специфически реагирующих лимфоцитов и развитием дисбаланса активности Th1 и Th2-субпопуляций [11], [13], [18], [30], [36]. В последнее время предпринимаются попытки объяснить накопление функционально активных Т-лимфоцитов с реактивностью в отношении собственных антигенов патологией программированной клеточной гибели.
Апоптоз как особая, генетически запрограммированная, форма гибели клетки является механизмом, ответственным в границах иммунной системы за элиминацию активированных лимфоцитов, выполнивших свою функцию, с целью предупреждения аутоиммунных реакций.
Механизмы апоптоза сложны и включают взаимодействие большой группы соединений, центральное место среди которых занимают особые протеазы – каспазы (К) (cytosolic aspartate-specific cysteine proteases), расщепляющие белки в местах расположения аспарагиновых оснований [4], [9], [29], [33]. По своим функциональным обязанностям и структурной гомологии К подразделяются на три группы: активаторы цитокинов (К1, К4, К5, К13), активаторы эффекторных К (К2, К8, К9, К10) и эффекторные К, или непосредственные исполнители апоптоза (К3, К6, К7) [4], [5], [19], [21]. К расположены в цитоплазме в неактивном состоянии для исключения возможности случайной гибели клетки и активируются главным образом в последовательных реакциях пути «рецепторов смерти» и митохондриального пути [5], [9], [21].
Путь «рецепторов смерти» запускается взаимодействием специфических рецепторов на поверхности клетки-мишени с их лигандами – экстрацеллюлярными «белками смерти», наиболее изученным из которых является Fas-лиганд (FasL) [2], [7], [8], [9]. Физиологически в имму-нопривилегированных тканях (тестикулы и ткани глаза) FasL обеспечивает защиту от повреждения собственных клеток, вызывая апоптоз лимфоцитов [20], [22], [25].
При связывании FasL со специфическим рецептором Fas образуется DISC (death-inducing signalling complex, индуцирующий смерть сигнальный комплекс), итогом формирования которого является активация протеазы – К8 (FLICE), обеспечивающей процесс апоптоза [5], [20], [25].
К8 запускает программированную клеточную гибель превращением проК3 в К3 или расщеплением белка Вid семейства Bcl-2 с индукцией митохондриального пути апоптоза [4], [21], [23], [25], [33]. К3 активирует прочие эффекторные К (К6, К7), вместе с которыми осуществляет расщепление структурных белков, фрагментацию и нарушение репарации ДНК [3], [4], [19].
Митохондриальный путь апоптоза инициируется повреждением ДНК или воздействием цитотоксических агентов [21]. Распознавание свободных концов ДНК сопровождается активацией гена р53, кодирующего образование ядерного протеина, который способен в случае большого количества повреждений запускать программу гибели клетки [3], [4], [7], [8], [33]. Помимо стимуляции экспрессии Fas и К6 через р53 отмечена активация генов индукторов апоптоза, относящихся к белкам семейства Bcl-2 [2], [29], [35].
Семейство Bcl-2 объединяет сходные по морфологическому составу белки (более 20 соединений), разделяющиеся на две группы в зависимости от функции: индукторы апоптоза (Bim, Bad, Bid, Bax, Bak, Bok) и ингибиторы апоптоза (Bcl-2, Bcl-w, А1, Mcl-1, Bcl-x L ) [2], [3], [4], [21]. Индукторы клеточной гибели семейства Bcl-2 (Bax, Bak и др.) способствуют выделению из митохондрий цитохрома с, который, взаимодействуя с Apaf-1 (apoptotic protease activating factor-1, апоптотиче-ский протеаза-активирующий фактор-1) и проК9, формирует комплекс, называемый апоптосомой. В апоптосоме проК9 превращается в К9, активирующую эффекторные К3, К6, К7 [5], [8], [19], [21], [25], [32]. Антиапоптозные белки семейства Bcl-2 связываются с белком Apaf-1 во время формирования апоптосомы, что предотвращает активацию каспазного каскада [2], [29]. Bcl-2 способен ингибировать высвобождение цитохрома с из митохондрий [7], [29].
Помимо соединений, индуцирующих апоптоз, в клетках существуют механизмы негативной регуляции этого процесса – эндогенные ингибиторы К (белки семейства IAP (inhibitor of apoptosis, ингибитор апоптоза)) [1], [2], [3], [19], [25] и структурные гомологи проксимальных К8 и К10 [19], [29], [33], [34].
К прочим митохондриальным факторам апоптоза, расположенным в межмембранном пространстве, относят фактор, индуцирующий апоптоз (AIF, apoptosis-inducing factor), который активирует нуклеазу, расщепляющую ядерную ДНК с конденсацией хроматина, запуская каспазонезависимую гибель клетки [4], [21], [29], [32], [33].
Исходя из предполагаемой роли апоптоза в патогенезе РА, в настоящее время активно изучается влияние базисных противовоспалительных препаратов (БПВП) на программированную клеточную гибель с целью объяснения их патогенетического действия, предсказания эффективности использования и разработки методов преодоления резистентности, которая остается одной из основных проблем в фармакотерапии РА.
В настоящее время препаратом первого ряда в лечении больных РА является метотрексат [10], [13], [14], [15], [18]. Основным механизмом действия метотрексата считается антифолатный с ингибированием образования пуринов и тимидилата, необходимых для синтеза ДНК, что ведет к антипролиферативному эффекту [10], [14], [18], [24]. Противовоспалительная активность связана с высвобождением аденозина, который, взаимодействуя с аденозиновыми рецепторами 2-го типа, ингибирует лимфоцитарную пролиферацию, образование лейкотриена В4 в нейтрофилах, продукцию фактора некроза опухоли-альфа (ФНО-α), интерлейкинов [10], [14], [18], [24], [28].
Показана способность метотрексата индуцировать апоптоз различных клеток, в том числе периферических лимфоцитов [14], [24], [26], [30], [31]. Несмотря на то что ранее продемонстрировано участие Fas в индукции метотрексатом апоптоза лейкозных клеток [14], [24], в последнее время все больше данных о независимости программированной клеточной гибели от системы Fas/FasL [27], [30], [31]. Так, апоптоз активированных Т-клеток здоровых доноров in vitro, индуцированный метотрексатом, не нарушался при добавлении ингибиторов CD95, полностью блокирующих Fas-опосредованную гибель [27], [30]. На мышиных моделях с дефектом Fas происходящая индукция апоптоза метотрексатом предполагала независимость от стимуляции CD95 [31].
Отмечена способность аденозина вызывать апоптоз в ограниченном числе Т-клеток с уменьшением этого эффекта применением ингибитора аденозиндезаминазы [27]. Не исключено, что метотрексат активирует программированную клеточную гибель опосредованно, влияя на метаболизм основных цитокинов при РА, выделяемых в большом количестве активированными иммунокомпетентными клетками [14], [24].
Индукция программированной клеточной гибели как возможный механизм действия описан и для сульфасалазина – еще одного БПВП, часто используемого в терапии РА. Сульфасалазин представляет собой конъюгат 5-аминосалици-ловой кислоты и сульфапиридина и является первым базисным препаратом, который разрабатывался для лечения РА, исходя из представлений об инфекционной природе заболевания [12]. Отсутствие влияния ингибитора К широкого спектра на индуцированный сульфасалазином апоптоз предполагает каспазонезависимый механизм программированной клеточной гибели [12], [32]. Инициирующим звеном в механизме запуска апоптоза считается белок Bax, который в ответ на терапию транслоцируется в митохондрии и вызывает высвобождение проапоптогенных факторов, в том числе AIF, с последующей фрагментацией ДНК [32]. Среди прочих механизмов влияния сульфасалазина на интенсивность апоптоза иммунокомпетентных клеток также описывается подавление экспрессии провоспалительных цитокинов (ФНО-α, интерлейкинов) [1].
Таким образом, нарушения апоптоза периферических лимфоцитов могут играть важную роль в развитии и хронизации иммунопатологического воспалительного процесса при РА. Активация программированной клеточной гибели при эффективной терапии БПВП способна подтвердить значение апоптоза в патогенезе заболевания, а понимание механизмов реализации действия базисных средств поможет объяснить различную чувствительность к ним пациентов.
Целью настоящего исследования явилось изучение динамики активности апоптоза периферических лимфоцитов на фоне проводимой базисной терапии, оценка возможности использования показателей активности программированной клеточной гибели для прогнозирования эффективности терапии БПВП.
МАТЕРИАЛЫ И МЕТОДЫ
В исследуемую группу включено 90 пациентов, из которых 63 (70 %) женщины и 27 (30 %) мужчин, в возрасте от 20 до 74 лет (средний возраст – 49,34 ± 11,63 года) с РА, диагностированным на основании критериев Американской коллегии ревматологов. У 87 (96,67 %) пациентов РА диагностирован впервые или в предшествующие исследованию 36 месяцев.
У всех больных РА проводилась клиническая оценка общего суставного синдрома (суставной индекс Ричи (СИР), число болезненных суставов (ЧБС), число припухших суставов (ЧПС), утренняя скованность (УС), оценка состояния здоровья и боли пациентом по визуальной аналоговой шкале (ВАШ)) и функционального статуса по вопроснику HAQ. Для определения стадии РА всем больным проводилась рентгенография клинически наиболее пораженных суставов. Общая активность РА оценивалась по индексу DAS4. Клиническая характеристика исследуемой группы представлена в табл. 1.
Пациентам выполнялось исследование гемограммы, определялся уровень общего белка и белковых фракций, фибриногена, С-реактивного белка с помощью высокочувствительного теста (hsСРБ), ревматоидного фактора (РФ), циркулирующих иммунных комплексов (ЦИК).
Оценка апоптоза циркулирующих лимфоцитов проводилась в лаборатории кафедры молекулярной биологии, органической и неорганической химии ГОУ ВПО «Петрозаводский государственный университет» по активности К4, К6 и К8 через 30, 60, 120, 150 и 180 минут после начала реакции флуоресценции (ФЦ) с использованием специфических субстратов К (для К4 (активатор цитокинов) – WEHD (Trp = Glu = His = Asp), для К8 (активатор эффекторных К) – LETD (Leu = Glu = Thr = Asp), для К6 (эффекторная К) – VEID (Val = Glu = Ile = Asp)) и по выявлению количества 2-нитевых и 1-нитевых разрывов ДНК методом ФЦ ДНК-тропных красителей – бромистого этидия (EtBr) (специфичен для выявления 2-нитевых разрывов ДНК) и 4’,6-диамидино-2-фенилиндола (DAPI) (специфичен для выявления 1-нитевых разрывов ДНК).
Клиническая характеристика исследуемой группы
|
Характеристика заболевания |
Число пациентов |
% больных |
|
Серопозитивный вариант |
59 |
65,56 |
|
Серонегативный вариант |
31 |
34,44 |
|
Системные проявления |
57 |
63,33 |
Рентгенологическая стадия
|
Нет изменений |
8 |
8,89 |
|
I |
5 |
5,56 |
|
II |
59 |
65,55 |
|
III |
15 |
16,67 |
|
IV |
3 |
3,33 |
|
Степень активности |
||
|
I |
23 |
25,56 |
|
II |
45 |
50,00 |
|
III |
22 |
24,44 |
|
Функциональная недостаточность |
||
|
0 |
13 |
14,44 |
|
I |
42 |
46,67 |
|
II |
34 |
37,78 |
|
III |
1 |
1,11 |
Все больные РА были разделены на 3 группы в зависимости от длительности заболевания и особенностей проводимого лечения. Группу впервые диагностированного РА (ВДРА) составили 25 (27,78 %) пациентов, которые были включены в исследование сразу после установления диагноза. Больные этой группы имели начальные клинические проявления заболевания длительностью менее 6 месяцев и никогда ранее не получали БПВП.
Группу пациентов с установленным ранее РА, не принимающих базисные препараты (БНБП) по разным причинам более 3 месяцев перед первичным осмотром, составили 26 (28,89 %) больных.
В группу пациентов с ранее диагностированным РА, получающих постоянную базисную терапию (БПБТ) на протяжении 3 и более месяцев перед исследованием, включено 39 (43,33 %) больных – в качестве базисного средства использовался главным образом метотрексат в дозе 7,5–20 мг/нед. (в среднем 11,82 ± 3,47 мг/нед.).
Группы были сопоставимы по полу, возрасту, показателям суставного синдрома, активности (DAS4), количеству серопозитивных пациентов. Для больных с установленным ранее РА отсутствовали различия по стадиям и длительности болезни. Общая характеристика групп представлена в табл. 2, клиническая характеристика – в табл. 3.
Рекомендованная после первичного обследования базисная терапия включала преимущественно метотрексат в дозе 7,5–20 мг/нед. (в среднем 11,13 ± 3,24 мг/нед.) – у 75 (83,33 %) пациентов, а также сульфасалазин в дозе 2 г/сут. – у 11 (12,22 %) больных.
Таблица 3
|
Клиническая характеристика |
||||||
|
групп пациентов |
||||||
|
Характеристика |
ВДРА |
БНБП |
БПБТ |
|||
|
группы |
(n = |
25) |
(n = |
26) |
(n = |
39) |
|
кол-во |
% |
кол-во |
% |
кол-во |
% |
|
|
Серопозитивный |
16 |
64,00 |
18 |
69,23 |
25 |
64,10 |
|
вариант Серонегативный вариант |
9 |
8 |
14 |
|||
|
36,00 |
30,77 |
35,90 |
||||
|
Рентгенологическая стадия |
||||||
|
Нет изменений |
7 |
28,00 |
0 |
0,00 |
1 |
2,56 |
|
I |
1 |
4,00 |
2 |
7,69 |
2 |
5,13 |
|
II |
14 |
56,00 |
17 |
65,38 |
28 |
71,80 |
|
III |
3 |
12,00 |
6 |
23,08 |
6 |
15,38 |
|
IV |
0 |
0,00 |
1 |
3,85 |
2 |
5,13 |
|
Степень активности |
||||||
|
I |
3 |
12,00 |
7 |
26,92 |
13 |
33,33 |
|
II |
17 |
68,00 |
12 |
46,16 |
16 |
41,03 |
|
III |
5 |
20,00 |
7 |
26,92 |
10 |
25,64 |
|
Функциональная недостаточность |
||||||
|
0 |
1 |
4,00 |
4 |
15,38 |
8 |
20,51 |
|
I |
14 |
56,00 |
14 |
53,85 |
14 |
35,90 |
|
II |
10 |
40,00 |
8 |
30,77 |
16 |
41,03 |
|
III |
0 |
0,00 |
0 |
0,00 |
1 |
2,56 |
Таблица 1
|
Таблица 2 Общая характеристика групп пациентов |
||||||
|
Характеристика группы |
ВДРА (n = 25) |
БНБП (n = 26) |
БПБТ (n = 39) |
|||
|
Возраст, годы |
50,96 ± 11,51 |
46,58 ± 14,21 |
50,15 ± 9,62 |
|||
|
Длительность РА, мес. |
20,77 ± 13,11 |
19,05 ± 10,73 |
||||
|
кол-во |
% |
кол-во |
% |
кол-во |
% |
|
|
Женщины |
18 |
72,00 |
17 |
65,38 |
28 |
71,79 |
|
Мужчины |
7 |
28,00 |
9 |
34,62 |
11 |
28,21 |
Статистическую обработку полученных данных проводили с использованием Microsoft Office Excel 2003 и пакета статистических программ Statistica 6.0. Результаты представлены в виде средних величин и стандартных отклонений (M ± s, где М – среднее арифметическое, s – стандартное отклонение), а также медианы и интерквартильного размаха (A (L-H), где A – медиана, L – нижний квартиль, H – верхний квартиль). Статистический анализ проводился непараметрическими методами – сравнение 2 независимых групп методом Манна – Уитни, анализ корреляции методом Спирмена.
РЕЗУЛЬТАТЫ
В исследуемой группе на фоне терапии БПВП в течение года отмечено достоверное снижение индекса DAS4 и улучшение функционального статуса при повышении количества 1-нитевых разрывов ДНК. Результаты отражены на рис. 1.
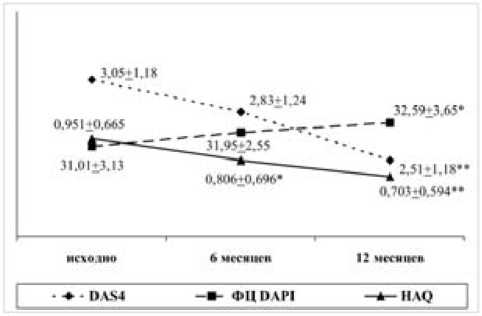
Рис. 1. Динамика индекса DAS4, HAQ и количества 1-нитевых разрывов ДНК в исследуемой группе на фоне терапии БПВП в течение 1 года
Достоверность различий при сравнении с исходными показателями отмечена * при p < 0,05, ** при p < 0,01.
Более интенсивная программированная клеточная гибель лимфоцитов может быть опосредована тенденцией к повышению активности К8 и К6 в течение 12 месяцев наблюдения (p > 0,05). Отмеченное снижение активности К4, участвующей в реализации провоспалительных процессов, на 180-й минуте от начала реакции ФЦ к 12 месяцам является достаточно закономерным с учетом снижения индекса DAS4.
При сравнительной оценке исходной активности апоптоза в группах ВДРА, БНБП и БПБТ с сопоставимыми показателями DAS4 количество разрывов ДНК достоверно не различалось. В группе ВДРА на фоне снижения DAS4 (p < 0,01) в течение года отмечена тенденция к увеличению количества 1-нитевых разрывов ДНК при достоверно более активных в динамике К6 и К8. Сохранялось закономерное снижение уровня К4, представленное на рис. 2.
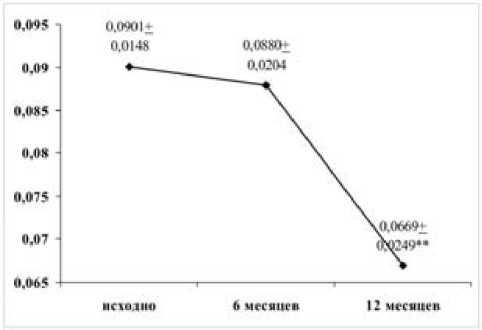
Рис. 2. Активность К4 в группе ВДРА на фоне терапии БПВП в течение 1 года
Достоверность различий при сравнении с исходными показателями отмечена * при p < 0,05, ** при p < 0,01.
В группе БНБП возобновленная или начатая базисная терапия также позволила достоверно снизить активность РА и улучшить функциональный статус пациентов в течение года (p < 0,05). Тенденция к увеличению количества 1-нитевых разрывов ДНК была статистически незначимой (p > 0,05), как и повышение активности К6, К8.
В группе БПБТ не продемонстрировано существенного изменения индекса DAS4 и показателей HAQ. Отмечена наименьшая динамика активности программированной клеточной гибели среди всех 3 групп при повышении активности К6 и К8, что может указывать на нарушения реализации апоптоза. Учитывая, что основную часть обследованных в группе БПБТ составляли пациенты стационара, можно предположить исходно недостаточный эффект рекомендованных базисных средств или развитие резистентности к ним, послужившие причиной очередной госпитализации.
При сравнении показателей активности апоптоза к 12 месяцам наблюдения в группах ВДРА, БНБП, БПБТ не отмечено достоверных различий по количеству разрывов ДНК.
Для оценки динамики активности апоптоза в зависимости от эффективности проводимой терапии больные с удовлетворительным или хорошим эффектом по критериям Европейской противоревматической лиги (EULAR), а также 20 %, 50 % и 70 % улучшением по критериям Американской коллегии ревматологов (ACR) составили группу «ответчиков». Пациенты, у которых была констатирована неэффективность лечения базисными средствами, вошли в группу «неответчиков».
Через 6 месяцев «ответчики» продемонстрировали тенденцию к снижению активности К4 и росту количества 1-нитевых разрывов ДНК (p > 0,05). В группе «неответчиков» к 6 месяцам при достоверном повышении DAS4 отмечено снижение уровня 2-нитевых разрывов ДНК (p < 0,05). Через 1 год наблюдения обращает внимание обратное изменение количества 1-нитевых разрывов ДНК и активности К4 (p < 0,05) у «не-ответчиков» на фоне более низких показателей DAS4. Не исключается, что определяемые изменения активности апоптоза реализуются улучшением клинической картины и лабораторных показателей только спустя некоторое время.
Для определения прогностической ценности результатов исследования апоптоза было проведено попарное сравнение исходных данных по активности К и количеству разрывов ДНК в группах «неответчиков» и «ответчиков» – достоверных различий выявлено не было.
При оценке влияния метотрексата на активность РА и интенсивность программированной клеточной гибели проведен анализ эффективности лечения у 23 пациентов групп ВДРА и БНБП, начавших терапию препаратом с момента включения в исследование и продолжавших ее в течение всего времени наблюдения без перерывов и смены базисного средства. Средняя реко- мендованная доза препарата при выписке из стационара исходно составила 9,46 ± 1,84 мг/нед., а к 12 месяцам – 11,67 ± 3,36 мг/нед. Динамика основных клинико-лабораторных данных представлена в табл. 4.
На фоне снижения индекса DAS4 и улучшения функционального статуса к 12 месяцам отмечена тенденция к повышению количества 1-нитевых разрывов ДНК, активности К6, К8 (p > 0,05), достоверное снижение активности К4 (p < 0,01).
Таблица 4
Динамика клинико-лабораторных показателей на фоне терапии метотрексатом в течение 1 года
|
Показатели |
Исходно |
Время от начала терапии метотрексатом |
|
|
6 месяцев |
12 месяцев |
||
|
DAS4 |
3,46 ± 0,91 |
2,77 ± 1,08 |
2,32 ± 1,10** |
|
HAQ |
1,266 ± 0,686 |
0,862 ± 0,659* |
0,725 ± 0,693* |
|
ЧБС |
6,0 (4,0–16,0) |
7,0 (1,0–12,0) |
4,0 (1,0–12,0) |
|
ЧПС |
9,0 (3,5–13,0) |
5,0 (2,0–9,0) |
2,0 (0,0–5,0)* |
|
СИР |
5,0 (4,0–11,0) |
5,0 (1,0–9,5) |
4,0 (1,0–8,5) |
|
ВАШ |
50,0 (43,5– |
43,0 (25,0– |
32,0 (8,0– |
|
боль, мм |
55,0) |
50,0) |
43,0)* |
|
ВАШ |
45,0 (39,0– |
45,0 (22,5– |
33,0 (8,0– |
|
активность, мм |
50,0) |
53,5) |
42,5)* |
|
УС, мин |
90,0 (42,5– |
30,0 (17,5– |
10,0 (0,0– |
|
165,0) |
150,0)* |
40,0)** |
|
|
hsСРБ, мг/л |
14,50 (3,47– |
5,96 (3,73– |
3,83 (1,86– |
|
35,35) |
11,20)* |
9,37) |
|
|
фибриноген, г/л |
5,5 (4,4–6,6) |
4,7 (3,7–5,7) |
4,3 (3,8–4,6)** |
|
α2-глобулин, г/л |
8,35 ± 1,50 |
6,43 ± 0,86** |
6,90 ± 0,83* |
|
РФ, МЕ/мл |
64 (16–256) |
64 (8–256) |
96 (32–256) |
|
ЦИК, ед. опт. |
0,135 ± 0,049 |
0,103 ± 0,041** |
0,128 ± 0,057 |
|
пл. |
|||
|
СОЭ, мм/ч |
30,0 (21,0– |
13,0 (10,0– |
14,0 (9,5– |
|
43,5) |
29,0)** |
24,0)** |
|
Примечание. Достоверность различий по сравнению с исходными показателями отмечена в таблице * при p < 0,05, ** при p < 0,01 и *** при p < 0,001.
ОБСУЖДЕНИЕ
Активное изучение особенностей программированной гибели клеток различных типов при РА обусловлено возможной ролью патологии апоптоза в развитии и поддержании ревматоидного воспаления. Влияние на процесс апоптоза считается одним из механизмов патогенетического действия БПВП. Исследование особенностей программированной клеточной гибели на фоне терапии БПВП может уточнить причины различной чувствительности пациентов к лекарственным средствам и оценить возможность ее прогнозирования.
В ходе работы выявлено, что терапия БПВП достоверно снижает активность РА, повышает качество жизни больных и сопровождается ростом интенсивности программированной клеточной гибели периферических лимфоцитов по количеству 1-нитевых разрывов ДНК, что может быть опосредовано более активными К8 и К6. Наиболее значимая динамика показателей активности апоптоза характерна для пациентов с ВДРА и БНБП при возобновлении лечения БПВП. Наименее эффективная терапия в группе БПБТ может объясняться исходно недостаточным действием БПВП и развитием резистентности к ним. Отсутствие достоверных различий к 12 месяцам по показателям программированной клеточной гибели в группах ВДРА, БНБП и БПБТ определяется небольшим количеством пациентов.
Динамика активности К4 в исследуемой группе соответствует динамике индекса DAS4 и отражает изменения активности РА на фоне лечения БПВП, что, наиболее вероятно, связано с ролью К4 как активатора цитокинов и участника провоспалительных реакций.
Результаты анализа не позволяют говорить о прогностической ценности показателей активности программированной клеточной гибели в отношении эффективности проводимой терапии БПВП, что требует дальнейшего изучения.
ЗАКЛЮЧЕНИЕ
Эффективное применение БПВП при РА сопровождается повышением активности апоптоза периферических лимфоцитов. Динамика активности К4 отражает интенсивность ревматоидного воспаления. Возможность использования показателей апоптоза периферических лимфоцитов для прогнозирования эффекта БПВП требует дальнейшего изучения.
СПИСОК СОКРАЩЕНИЙ
БНБП – больные, не получающие базисные препараты 3 месяца и более перед включением в исследование.
БПБТ – больные, получающие базисную терапию 3 месяца и более перед включением в исследование.
БПВП – базисные противовоспалительные препараты.
ВДРА – впервые диагностированный ревматоидный артрит.
К – каспаза.
СИР – суставной индекс Ричи.
РА – ревматоидный артрит.
РФ – ревматоидный фактор.
УС – утренняя скованность.
ФНО-α – фактор некроза опухоли-альфа.
ФЦ – флуоресценция.
ЦИК – циркулирующие иммунные комплексы.
ЧБС – число болезненных суставов.
ЧПС – число припухших суставов.
AIF – фактор, индуцирующий апоптоз.
Apaf-1 – апоптотический протеаза-активирующий фактор-1.
DAPI – 4’,6-диамидино-2-фенилиндол.
DISC – индуцирующий смерть сигнальный комплекс.
EtBr – бромистый этидий.
FasL – Fas-лиганд.
hsСРБ – высокочувствительный С-реактивный белок.
Список литературы Влияние базисной терапии ревматоидного артрита на активность апоптоза периферических лимфоцитов
- Бадокин В. В. Эффективность и переносимость сульфасалазина при ревматических заболеваниях//Научно-практич. ревматол. 2005. № 4. С. 47-52.
- Богданов А. Н., Камилова Т. А., Цыган В. Н., Цыган Е. Н. Роль апоптоза в патогенезе ревматоидного артрита. Сообщение 1//Научно-практич. ревматол. 2005. № 6. С. 56-62.
- Владимирская Е. Б. Биологические основы противоопухолевой терапии. М.: Агат-Мед, 2001.
- Владимирская Е. Б. Механизмы апоптотической смерти клеток//Гематология и трансфузиология. 2002. № 47 (2). С. 35-40.
- Григорьев М. Ю., Имянитов Е. Н., Хансон К. П. Апоптоз в норме и патологии//Мед. академ. журн. 2003. № 3 (3). С. 3-11.
- Коршунов Н. И. Ревматоидный артрит: диагностика и лечение//РМЖ. 2005. № 13 (14). С. 956-965.
- Мойбенко А. А., Досенко В. Е., Нагибин В. С. Ферментативные механизмы апоптоза//Патологическая физиология и экспериментальная терапия. 2005. № 3. С. 17-26.
- Москалева Е. Ю., Северин С. Е. Возможные механизмы адаптации клетки к повреждениям, индуцирующим программированную гибель. Связь с патологией//Патологическая физиология и экспериментальная терапия. 2006. № 2. С. 2-16.
- Нагорнев В. А., Восканьянц А. Н. Апоптоз и его роль в атерогенезе//Мед. академ. журн. 2003. № 3 (4). С. 3-18.
- Насонов Е. Л. 50 лет применения метотрексата в ревматологии//РМЖ. 2000. № 8 (9). С. 372-377.
- Насонов Е. Л. Фактор некроза опухоли-α -новая мишень для противовоспалительной терапии ревматоидного артрита//РМЖ. 2000. № 8 (17). С. 718-722.
- Насонов Е. Л. Применение сульфасалазина в ревматологии//Consilium-medicum. 2002. № 4 (8). С. 426-429.
- Насонов Е. Л. Фармакотерапия ревматоидного артрита с позиций доказательной медицины: новые рекомендации//РМЖ. 2002. № 10 (6). С. 294-302.
- Насонов Е. Л. Метотрексат: Перспективы применения в ревматологии. М.: Филоматис, 2005.
- Насонов Е. Л. Фармакотерапия ревматоидного артрита -взгляд в XXI век//Клин. мед. 2005. № 6. С. 8-12.
- Насонов Е. Л. Перспективы фармакотерапии ревматоидного артрита//Научно-практич. ревматол. 2005. № 6. С. 5-7.
- Насонова В. А. Ревматические болезни. М.: Медицина, 1997.
- Сигидин Я. А., Гусева Н. Г., Иванова М. М. Диффузные болезни соединительной ткани (системные ревматические заболевания): руководство для врачей. М.: Медицина, 2004.
- Фильченков А. А. Каспазы: регуляторы апоптоза и других клеточных функций//Биохимия. 2003. № 63 (4). С. 453-466.
- Червякова Н. В. Fas/Fas-лиганд: маркеры апоптоза//Лаборатория. 2004. № 2. С. 7-9.
- Adams J. M. Ways of dying: multiple pathways to apoptosis//Genes Dev. 2003. № 17. P. 2481-2495.
- Bohana -Kashtan O., Civin C. I. Fas Ligand as a tool for immunosupression and generation of immune tolerance//Stem Cells. 2004. № 22. P. 908-924.
- Breedveld F. C., Kalden J. R. Appropriate and effective management of rheumatoid arthritis//Ann. Rheum. Dis. 2004. № 63. P. 627-633.
- Cutolo M., Sulli A., Pizzorni C. et al. Anti-inflammatory mechanisms of methotrexate in rheumatoid arthritis//Ann. Rheum. Dis. 2001. № 60. P. 729-735.
- Eguchi K. Apoptosis in autoimmune disease//Intern. Med. 2001. № 40. Р. 275-284.
- Fairbanks L. D., Ruckemann K., Qui Y. et al. Methotrexate inhibits the first committed step of purine biosynthesis in mitogen-stimulated human T-lymphocytes: a metabolic basis for efficacy in rheumatoid arthritis?//Biochem. J. 1999. № 342. Р. 143-152.
- Genestier L., Paillot R., Fournel S. et al. Immunosuppressive properties of methotrexate: apoptosis and clonal deletion of activated peripheral T cells//J. Clin. Invest. 1998. № 102 (2). Р. 322-328.
- Gerards A. H., de Lathouder S., de Groot E. R. et al. Inhibition of cytokine production by methotrexate. Studies in healthy volunteers and patients with rheumatoid arthritis//Rheumatology. 2003. № 42. Р. 1189-1196.
- Haunstetter A., Izumo S. Apoptosis: basic mechanisms and implications for cardiovascular disease//Circ. Res. 1998. № 82. Р. 1111-1129.
- Herman S., Zurgil N., Langevitz P. et al. The induction of apoptosis by methotrexate in activated lymphocytes as indicated by fluorescence hyperpolarization: a possible model for predicting methotrexate therapy for rheumatoid arthritis patients//Cell Struct. Funct. 2003. № 28. Р. 113-122.
- Izeradjene K., Revillard J. -P., Genestier L. Inhibition of thymidine synthesis by folate analogues induces a Fas-Fas ligand-independent deletion of superantigen-reactive peripheral T cells//Int. Immunol. 2001. № 13 (1). P. 85-93.
- Liptay S., Fulda S., Schanbacher M. et al. Molecular mechanisms of sulfasalazine-induced T-cell apoptosis//Br. J. Pharmacol. 2002. № 137. Р. 608-620.
- Perlman H., Pagliari L. J., Volin M. V. Regulation of apoptosis and cell cycle activity in rheumatoid arthritis//Curr. Mol. Med. 2001. № 1. Р. 597-608.
- Pope R. M. Apoptosis as a therapeutic tool in rheumatoid arthritis//Nat. Rev. Immunol. 2002. № 2. Р. 1-9.
- Stahnke K., Fulda S., Friesen C. et al. Activation of apoptosis pathways in peripheral blood lymphocytes by in vivo chemotherapy//Blood. 2001. № 98. Р. 3066-3078.
- Weyand C. M. New insights into the pathogenesis of rheumatoid arthritis//Rheumatology. 2000. № 39 (suppl. 1). Р. 3-8.
