Влияние ожирения на развитие и прогрессию злокачественных новообразований: обзор современных данных и новых терапевтических мишеней

Автор: Семина Е.В., Данилова Н.В., Олейникова Н.А., Агапов М.А., Рубина К.А.
Журнал: Сибирский онкологический журнал @siboncoj
Рубрика: Обзоры
Статья в выпуске: 4 т.20, 2021 года.
Бесплатный доступ
Актуальность. Известно, что диабет 2-го типа, синдром обструктивного апноэ сна, остеоартроз и ряд злокачественных новообразований коррелируют с ожирением. Механизмы, обусловливающие взаимосвязь метаболических нарушений с возникновением злокачественных опухолей, пока неизвестны; значительную роль может играть изменение чувствительности к инсулину и факторам роста, изменение спектра секретируемых адипокинов или особенности их взаимодействия с рецепторами, изменение содержания стероидных половых гормонов в организме, а также особенности метаболизма глюкозы в опухолевых клетках (так называемый эффект Варбурга). Материал и методы. Проведен поиск источников, включая научные, клинические и обзорные статьи, опубликованные в рецензируемых журналах, индексируемых в pubmed, Wos, scopus и РИНЦ. Проанализировано более 150 статей, посвященных изучению взаимосвязи метаболических нарушений с опухолевой прогрессией, из которых 69 включены в данный обзор. Результаты. Основная стратегия лечения опухолевых заболеваний заключается в подавлении пролиферации опухолевых клеток и процессов метастазирования. Возможно, что значительную роль в этих процессах играет ряд факторов, способных усиливать побочные эффекты противоопухолевой терапии, поддерживать резистентность опухолевых клеток или изменять их метаболический профиль. Имеются новые данные о функции таких белков, как Т-кадгерин и рецептор урокиназы (upaR), и их возможном участии в регуляции метаболизма опухолевых клеток, в частности чувствительности к инсулину и гормонам жировой ткани. Помимо адипокинов как уже известных посредников, участвующих в канцерогенезе, в обзоре описаны новые данные о роли Т-кадгерина и урокиназного рецептора upaR в регуляции обменных процессов. Предложена модель, объясняющая взаимосвязь этих белков и метаболических нарушений, ассоциированных с процессами канцерогенеза и химиорезистентности опухолевых клеток. Заключение. Понимание факторов и механизмов, поддерживающих ожирение и нарушения метаболизма, с точки зрения их роли в канцерогенезе актуально как для разработки профилактических мер, так и для оптимизации терапевтических стратегий борьбы с опухолевыми заболеваниями.
Метаболический синдром, ожирение, инсулин, инсулинорезистентность, адипонектин, Т-кадгерин, рецептор урокиназы
Короткий адрес: https://sciup.org/140254543
IDR: 140254543 | УДК: 616-006.04-036:616-056.52 | DOI: 10.21294/1814-4861-2021-20-4-130-145
the relationship between metabolic disorders and tumor progression: review of present data and new therapeutic targets
Background. Type 2 diabetes mellitus, obstructive sleep apnea, osteoarthritis and certain types of cancer are known to correlate with obesity. The mechanisms underlying the link between metabolic disorders and cancer remain obscure, yet assuming a potentially important role of reduced insulin sensitivity, altered glucose metabolism in tumor cells (the so-called Warburg effect), changes in the spectrum of secreted adipokines or interaction with their cognitive receptors as well as changes in steroid sex hormone production. Material and methods. A search for articles published in peer-reviewed journals indexed in pubmed, Wos, scopus and Rsci was carried out. More than 150 articles devoted to the study of the relationship between metabolic disorders and tumor progression were analyzed, of which 69 were included in this review. Results. The main strategy of anticancer therapy is to suppress the proliferation of tumor cells and metastasis. However, one should take into consideration a significant role of additional factors that can enhance side effects of anticancer therapy, ensure the resistance of tumor cells to chemotherapy or change cancer cell metabolic profile. New data recently emerging in the literature indicate an important function of proteins such as t-cadherin and urokinase receptor (upar) and their possible involvement in the regulation of tumor cell metabolism, in particular, sensitivity to insulin and adipose tissue hormones. The review encompasses recent data on the involvement of t-cadherin and upar in the regulation of metabolism and proposes a model explaining the relationship between these proteins and metabolic disorders associated with the processes of carcinogenesis and chemoresistance of cancer cells. Conclusion. Understanding of the factors and mechanisms that support obesity and metabolic disorders is relevant both for the development of cancer preventive measures and optimization of therapeutic strategies for combating cancer.
Текст научной статьи Влияние ожирения на развитие и прогрессию злокачественных новообразований: обзор современных данных и новых терапевтических мишеней
Механизмы, связывающие ожирение с развитием и прогрессированием новообразований, включают развитие инсулинорезистентности и нарушение функционирования системы внутриклеточной сигнализации от инсулиноподобного фактора роста IGF-I; нарушение синтеза и метаболизма половых гормонов в организме; хроническое системное воспаление, вызываемое ожирением и поддерживаемое воспалительными цитокинами, которые вырабатывает жировая ткань; изменение уровня адипокинов – гормонов, продуцируемых жировой тканью [3, 4] (рис. 1).
Помимо способности к накоплению жира, жировая ткань является крупнейшим эндокринным органом, регулирующим энергетический баланс и обмен веществ, воспаление, иммунный ответ и другие функции [5]. Клетки жировой ткани осуществляют биосинтез и секрецию более 50 гормонов и цитокинов, известных в литературе как адипоцитокины, или адипокины.
Не менее важным явлением, ассоциированным с опухолевой прогрессией и метаболическими нарушениями, является кахексия, характеризуемая избыточным катаболизмом, повышенными затра- тами энергии и воспалением в различных системах органов, включая мышцы, жир и ЦНС. Раковая кахексия вызывает воспаление и дисфункцию жировой ткани, приводя к нарушению регуляции синтеза и секреции ряда провоспалительных и противовоспалительных адипокинов [6]. До сих пор механизмы влияния адипокинов при раковой кахексии до конца не ясны, а большинство исследований направлены на определение корреляции уровня адипокинов с опухолями, в то время как вариации синтеза, секреции и внутриклеточной сигнализации адипокинов, участвующих в ремоделировании жировой ткани при раковой кахексии, остаются не в фокусе внимания исследователей.
Одним из наиболее распространенных ади-покинов жировой ткани является адипонектин, который обладает инсулин-сенсибилизирующими, противовоспалительными и антиатерогенными свойствами [5]. Снижение уровня адипонектина в сыворотке ассоциировано не только с эндокринными расстройствами, инсулинорезистентностью, диабетом 2-го типа, атеросклерозом и ишемической болезнью сердца, но и со злокачественными новообразованиями. Адипонектин является ключевым медиатором в развитии и прогрессии некоторых видов злокачественных опухолей [7], однако механизмы взаимосвязи между ожирением и процессами канцерогенеза до сих пор недостаточно изучены. Недавно опубликованные данные указывают на важную роль и других адипоки-нов – лептина, резистина, висфатина, апелина и адипсина – в опухолевой прогрессии. Функции и свойства лептина изучены достаточно хорошо, а
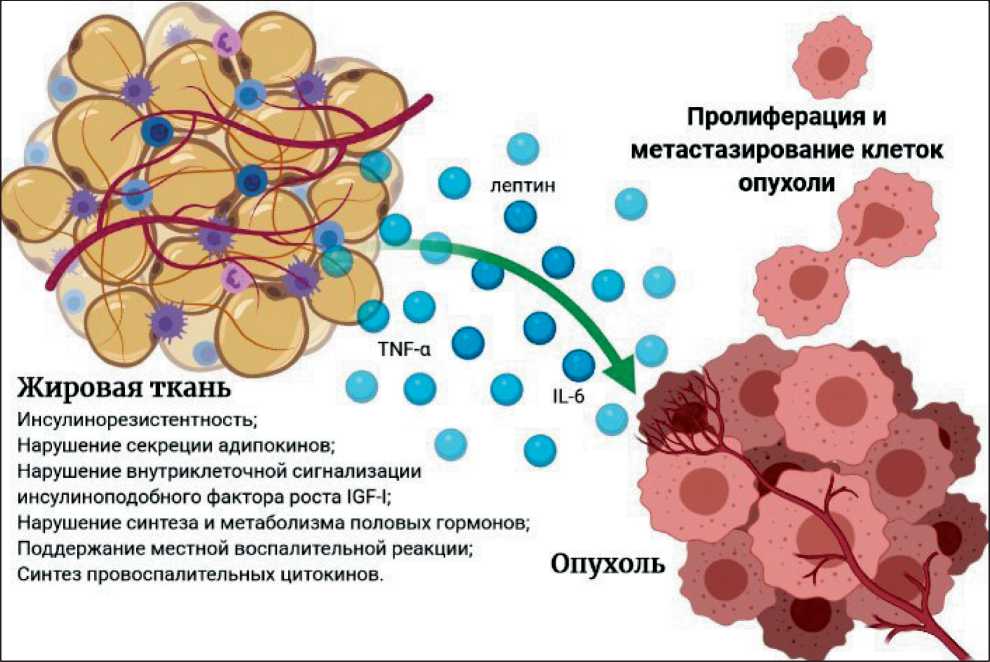
Рис. 1. Общие механизмы влияния ожирения на развитие и прогрессию новообразований .
Примечание: IGF-1 – инсулиноподобный фактор роста 1; IL-6 – интерлейкин 6; TNF-α – фактор некроза опухоли альфа Fig. 1. General mechanisms of the influence of obesity on the development and progression of cancer.
Note: IGF-1 – insulin-like growth factor 1, IL-6 – interleukin 6, TNF-α – tumor necrosis factor alpha
роль остальных четырех адипокинов в опухолевой прогрессии не определена, что требует дальнейших исследований.
Взаимосвязь ожирения с риском развития опухолевого заболевания и его прогрессией
В некоторых исследованиях обнаружено, что избыточная масса тела увеличивает риск возникновения рака молочной железы (РМЖ) на 30–50 % у женщин в постменопаузе, кроме того, центральное ожирение является независимым прогностическим фактором риска развития РМЖ в постменопаузе. Анализ исходов РМЖ показал, что ожирение связано как со снижением выживаемости, так и с повышением вероятности рецидива, независимо от менопаузального статуса и исходной стадии рака. У женщин с ожирением III степени (ИМТ=40,0 кг/м2) смертность от рака молочной железы была в 2 раза выше, чем у худых пациенток (ИМТ<20,5 кг/м2) [10].
Избыточный вес и ожирение увеличивают риск развития рака эндометрия (относительный риск 1,6 при увеличении ИМТ на 5 кг/м2) [11], однако в литературе встречаются и другие данные: у тучных женщин при опухолях I стадии прогноз более благоприятный, чем при II стадии. Предполагается, что такие противоречия обусловлены молекулярной неоднородностью ткани опухоли, и ожирение связано с менее агрессивными молекулярными подтипами опухолевых клеток.
Риск возникновения почечно-клеточного рака также оказывается выше у людей с избыточным весом и ожирением, чем с нормальным весом (относительный риск 1,2–1,3 при увеличении ИМТ на 5 кг/м2) [11]. Несмотря на то, что и ожирение, и артериальная гипертензия являются проявлениями МС, риск почечно-клеточного рака ассоциирован только с ожирением, но не с артериальным давлением. Скорее всего, при раке почки ожирение ассоциировано с более индолентными молекулярными вариантами (в частности, со снижением экспрессии синтазы жирных кислот FASN, нарушением регуляции в экспрессии ряда генов и т. д.), в то время как при раке яичников, напротив, высокий ИМТ ассоциирован с неблагоприятным прогнозом (низкодифференцированный серозный и эндометриоидный подтипы рака), и при этих гистологических подтипах существует линейная положительная связь между ИМТ и риском смертности, которая отсутствует при высокодифференцированном серозном раке яичников.
Несмотря на то, что высокий ИМТ является адекватным показателем избыточного веса и ожирения в клинических исследованиях, он не всегда отражает вызываемое метаболическими расстройствами ожирение, которое может быть ассоциировано с канцерогенезом. Показано, что 25 % людей, страдающих ожирением, метаболически здоровы, а имеющие ИМТ в пределах нормы имеют нарушения метаболизма [12].
Ниже рассмотрены известные на сегодняшний момент данные о молекулярных механизмах взаимосвязи метаболических нарушений, вызванных ожирением, с риском возникновения опухолевых заболеваний: инсулинорезистентность, изменение продукции адипокинов и половых гормонов, ремоделирование матрикса системой активаторов плазминогена, а также воспаление (рис. 2).
Инсулинорезистентность
Инсулинорезистентность и система сигнализации от инсулиноподобного фактора роста 1 (ИФР-1) частично опосредуют взаимосвязь между ожирением и раком. При инсулинорезистентности, часто наблюдаемой у пациентов с ожирением, уровень инсулина в плазме крови часто оказывается повышен, что является компенсаторной реакцией, направленной на предотвращение гипергликемии. Инсулин воздействует на рецепторы соматотропного гормона (СТГ) в печени и стимулирует в ней выработку ИФР-1 [13]. Можно было бы ожидать, что уровень ИФР-1 в сыворотке крови коррелирует с ИМТ, однако уровень ИФР-1 у пациентов с ожирением остается нормальным или даже сниженным. Частично это может быть объяснено ингибирующим действием высоких концентраций инсулина на секрецию белка, связывающего ИФР-1/2, а следующее за этим увеличение уровня свободного ИФР-1 по принципу обратной связи приводит к снижению секреции СТГ, что в конечном итоге вызывает понижение концентрации ИФР-1 в крови [13].
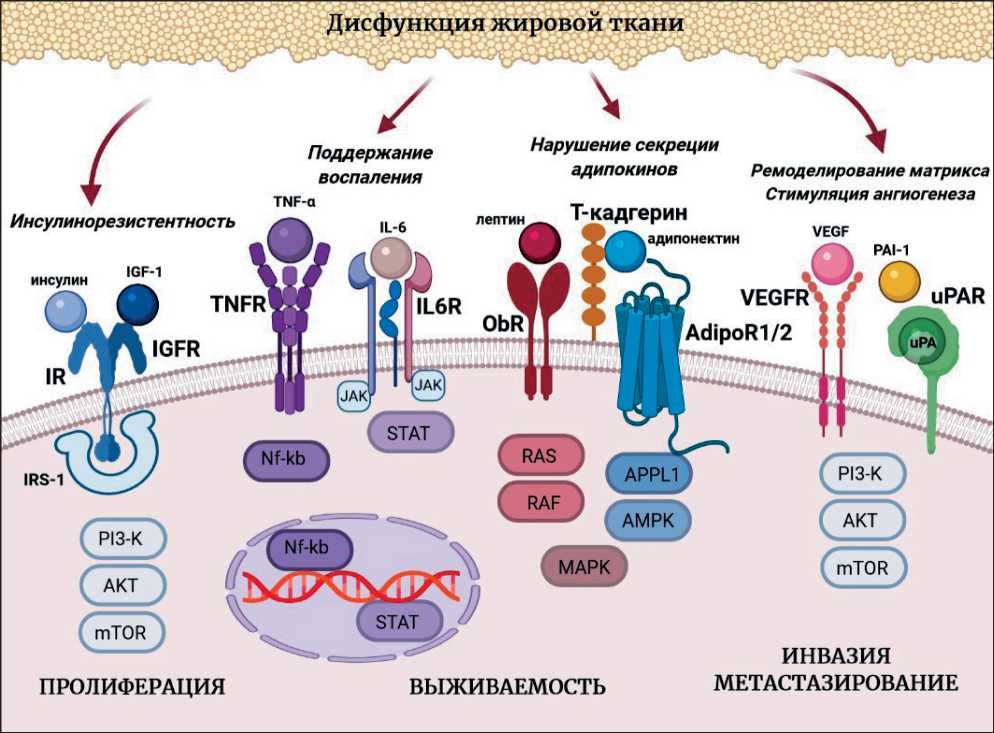
Рис. 2. Потенциальные сигнальные пути, напрямую связывающие ожирение с опухолью.
Примечание: AdipoR1/2 – рецептор адипонектина 1/2; AKT – протеинкиназа B; AMPK – 5'АМФ-активируемая протеинкиназа; APPl1 – адаптерный белок, фосфотирозин; IGF-1 – инсулиноподобный фактор роста 1; IGFR – рецептор инсулиноподобного фактора роста 1; IL-6 – интерлейкин-6; IL6R – рецептор интерлейкина-6; IR – инсулиновый рецептор;
IRS-1 – субстрат 1 рецептора инсулина; JAK – янус-киназа 1, MAPK – митоген-активируемая протеинкиназа;
mTOR – мишень рапамицина млекопитающих; NF-kb – ядерный фактор kb; ObR – рецептор лептина, PAI-1 – ингибитор активатора плазминогена 1; PI3-K – фосфоинозитид-3-киназа; RAF – серин/треониновая протеинкиназа; RAS – малые ГТФазы; STAT – сигнальный трансдуктор и активатор транскрипции; TNF-α – фактор некроза опухоли α; TNFR – рецептор фактора некроза опухоли; uPA – урокиназа, uPAR – рецептор урокиназы; VEGF – фактор роста эндотелия сосудов;
VEGFR – рецептор фактора роста эндотелия сосудов
Fig. 2. Potential signaling pathways directly linking obesity to tumor. Note: AdipoR1/2 – adiponectin 1/2 receptor, AKT – protein kinase B, AMPK – 5'AMP-activated protein kinase, APPl1 – adapter protein, phosphotyrosine, IGF-1 – insulin-like growth factor 1, IGFR – insulin-like growth factor receptor 1, IL-6 – interleukin-6, IL6R – interleukin-6 receptor, IR – insulin receptor, IRS-1 – insulin receptor substrate 1, JAK – Janus kinase 1, MAPK – mitogen-activated protein kinase, mTOR – mammalian rapamycin target, NF-kb – nuclear factor kb, ObR – leptin receptor, PAI-1 – plasminogen activator 1 inhibitor, PI3-K – phosphoinositide 3-kinase, RAF – serine / threonine protein kinase, RAS – small GTPases, STAT – signal transducer and transcriptional activator, TNF-α – tumor necrosis factor α, TNFR – tumor necrosis factor receptor, uPA – urokinase, uPAR – urokinase receptor, VEGF – vascular endothelial growth factor, VEGFR – vascular endothelial growth factor receptor
Предполагается, что и инсулин, и ИФР-1 играют важную роль в опухолевой прогрессии за счет связывания с рецептором инсулина (IR) и рецептором ИФР-1 соответственно. ИФР-1 может ингибировать апоптоз и стимулировать пролиферацию клеток за счет активации нескольких сигнальных путей, включая систему фосфатидилинозитол-3-киназы (PI3Ks) и систему Ras/Raf/митоген-активируемых протеинкиназ (MAPK), а также сигнализацию с участием mTOR и белков-регуляторов апоптоза Bad/Bcl2. Экспрессия ИФР-1 и его рецептора увеличивается в ряде опухолей (КРР, раке простаты, молочной железы, яичника и легкого, глиобластоме, нейробластоме), что позволяет предполагать, что ИФР-1 может оказывать стимулирующее действие на опухолевый рост по принципу положительной обратной связи. В клетках карциномы поджелудочной железы человека ИФР-1 стимулирует миграцию и инвазию путем индукции экспрессии активатора плазминогена урокиназного типа (урокиназы, uPA) и его рецептора (uPAR), которые обеспечивают деградацию внеклеточного матрикса за счет активности урокиназы, связанной с рецептором, и активации внутриклеточной сигнализации [14].
Помимо регуляции транспорта глюкозы, инсулин обладает митогенным и антиапоптотическим действием, которое реализуется через схожую с ИФР-1 внутриклеточную сигнализацию, рекрутируя рецепторы факторов роста и активируя специфическую сигнализацию с участием Sos-Ras-Raf-Map сигнального каскада. В условиях повышенного содержания инсулина и ИФР-1 в сыворотке крови создаются митогенные и антиапоп-тотические условия, ускоряющие пролиферацию клеток и накопление мутаций, что способствует канцерогенезу. Клинические исследования показали, что пациенты с высоким уровнем ИФР-1 имеют повышенный риск развития таких видов рака, как колоректальный рак, рак простаты и молочной железы. Показано, что гиперинсулинемия является независимым фактором риска развития рака молочной железы и повышает риск возникновения КРР и рака эндометрия [15]. Кроме того, сахарный диабет, сопровождаемый инсулинорезистентно-стью, связан с повышенным риском возникновения РМЖ, КРР, рака поджелудочной железы и мочевого пузыря. Резистентность к инсулину играет важную роль в канцерогенезе и является одним из основных механизмов, обусловливающих взаимосвязь метаболических нарушений, ожирения и опухолевой прогрессии.
Адипокины и адипонектин
Жировая ткань вырабатывает широкий спектр гормонов и цитокинов, известных как адипокины. В эту группу входят такие основные белки, как адипонектин, резистин, висфатин, апелин, адипсин; дисфункция жировой ткани приводит к изменению секреции адипокинов в кровь, что непосредственно влияет на процессы ожирения и ассоциированный с ним злокачественный рост [16].
Адипонектин продуцируется белыми адипоцитами и циркулирует в крови в высоких концентрациях (до 0,01 % общего белка плазмы крови). Установлено, что концентрация адипонектина в плазме крови снижается при ожирении. Системное введение высокомолекулярного адипонектина обладает инсулин-сенситизирующим, антиатероген-ным, противовоспалительным и, в определенных условиях, снижающим ИМТ эффектами у человека и у мыши. Адипонектин обнаруживается в крови в виде форм с различной молекулярной массой: тримеры с низким молекулярным весом (LMW, low molecular weight) – гексамеров (MMW, medium molecular weight), олигомеров с высоким молекулярным весом (HMW, high molecular weight), состоящих из 4–6 тримеров. Именно высокомолекулярные комплексы (HMW) адипонектина оказывают стимулирующее действие на метаболизм.
Достаточно давно были идентифицированы два рецептора адипонектина, AdipoR1 и AdipoR2, которые широко экспрессируются в органах и тканях всего организма с преимущественной экспрессией AdipoR1 в печени, скелетных мышцах, макрофагах, гипоталамусе и ряде других органов и активацией сигнализации с участием 5'АМФ-активируемой протеинкиназы AMPK; и AdipoR2, секретируемым печенью, белыми адипоцитами, сосудистыми клетками и с активацией PPAR. Считается, что адипонектин, связываясь с рецепторами AdipoR1, AdipoR2, стимулирует окисление жирных кислот, увеличивает чувствительность клеток к инсулину и утилизацию глюкозы. Данные литературы свидетельствуют о том, что при связывании адипо-нектина с AdipoR1 AdipoR2 происходит рекрутирование адапторного белка APPL1, что приводит к целому спектру сигнальных эффектов, а именно к активации сигнализации с участием AMPK, mTOR,
PI3K/AKT, MAPK, STAT3 и NF-kB [17]. Одним из механизмов, лежащих в основе положительных эффектов адипонектина, является активация AMPK (5'АМФ-активируемая протеинкиназа). AMPK представляет собой белковый комплекс, состоящий из каталитической α субъединицы и двух регуляторных β и γ субъединиц. АМРК является ключевым регулятором энергетического баланса клетки, поскольку активируется при интенсивных физических нагрузках, стимулирует катаболические процессы и регулирует соотношение АМФ и АТФ в клетках, а также подавляет энергозатратные процессы глюконеогенеза и липогенеза [18].
Помимо остановки деления клеток опухоли, адипонектин запускает апоптоз путем стимуляции ключевых белков регуляторов клеточного цикла р53 и р21. Кроме того, описаны как минимум ещё 2 механизма прямого антиканцерогенного действия адипонектина: активированная AMPK напрямую фосфорилирует опухолевый супрессор туберин или комплекс туберозного склероза 2 (TSC2); ингибирует mTOR (механистическая мишень рапамицина).
Независимо от активации AMPK адипонектин снижает выработку активных форм кислорода, что может приводить к снижению активации MAPK киназ, следовательно, как и в случае фосфорилирования TSC2 и ингибирования mTOR, подавляет пролиферацию клеток. In vitro адипонектин ингибирует рост нескольких клеточных линий рака молочной железы и вызывает апоптоз клеток миеломоноцитарного происхождения (лейкемии). Также было показано, что адипонектин ингибирует опухолевый неоангиогенез. Этот эффект адипонектина частично опосредован активацией каскада каспаз (каспазы -8, -9 и -3), которые вызывают апоптоз эндотелиальных клеток. Ряд исследований, проведенных на трансгенных мышах A-ZIP/F-1 (мыши, у которых отсутствует белый жир), показал, что резистентность к инсулину и воспаление, наряду с адипокинами, играют важную роль в регуляции роста клеток рака молочной железа. У мышей A-ZIP/F-1, у которых отсутствует секреция адипокинов, чаще развивается диабет и регистрируется повышенный статус воспаления; в целом, эти мыши более восприимчивы к канцероген-индуцированному формированию и росту опухолей, чем мыши дикого типа [19]. Ускоренное формирование опухолей у таких мышей в отсутствие продукции адипокинов подтверждает противоопухолевые свойства адипокинов и прежде всего адипонектина.
Имеется противоречивая информация о взаимосвязи раковой кахексии и секреции адипонек-тина. В некоторых исследованиях, в том числе с использованием животных моделей кахексии, показано, что уровень адипонектина в сыворотке существенно возрастал уже на ранней стадии кахексии, а к более поздней стадии значительно снижался. Возможно, что такая динамика адипо-нектина зависит от области его секреции: в брыжеечном жиру экспрессия гена адипонектина по мере прогрессии кахексии возрастала, в то время как в забрюшинном и эпидидимальном жировых депо, наоборот, снижалась [20].
Т-кадгерин – рецептор адипонектина
Недавно был обнаружен третий рецептор высокомолекулярного (HMW) адипонектина – T-кадгерин. У человека и у мыши Т-кадгерин экспрессируется в нервной системе, сердце, сосудах (в эндотелии, перицитах и гладкомышечных клетках), и его экспрессия повышается при таких патологических состояниях, как атеросклероз и рестеноз [21].
Наша лаборатория в течение последних 25 лет занималась поисками рецепторов, опосредующих негативные сигнальные эффекты липопротеидов, и в качестве такого рецептора нами был идентифицирован Т-кадгерин [22]. Была сформулирована гипотеза о конкуренции двух лигандов, липопротеинов низкой плотности (ЛНП) и ади-понектина, за связывание с Т-кадгерином [23], согласно которой в здоровом организме T-кадгерин преимущественно оккупирован адипонектином, а при снижении концентрации адипонектина в условиях МС Т-кадгерин оказывается связан с ЛНП. Экспрессия мРНК всех трех рецепторов (AdipoR1, AdipoR2 и Т-кадгерина) тесно взаимосвязана между собой и коррелирует с экспрессией PPARδ, который является важнейшим регулятором функционирования жировой ткани, так как повышает дифференцировку адипоцитов и увеличивает количество мелких адипоцитов, чувствительных к инсулину. В мышечной ткани уровень экспрессии каждого из трех рецепторов изменяется и обратно коррелирует с уровнем триглицеридов, что предполагает регуляцию экспрессии Т-кадгерина и
AdipoR1/AdipoR1 в зависимости от метаболического статуса, а также то, что, наряду с AdipoR1 и AdipoR2, Т-кадгерин может участвовать в активации внутриклеточной сигнализации, описанной выше для адипонектина [24].
Генетические исследования однонуклеотидных полиморфизмов человека (Single Nucleotide Polymorphisms – SNPs) показали, что полиморфизмы в гене Т-кадгерина ( rs4783244 и rs12051272 ) влияют на уровень экспрессии адипонектина. У мышей, нокаутных по Т-кадгерину, уровень адипонектина в эндотелиальных клетках, сердце и мышцах достоверно снижен, а в крови повышен, что отражает неспособность адипонектина фиксироваться в сердце и сосудах в отсутствие Т-кадгерина [25]. Данные, полученные на экспериментальной модели инфаркта у мышей, нока-утных по Т-кадгерину, показали, что отсутствие Т-кадгерина или адипонектина в равной степени приводит к развитию гипертрофии сердца и увеличению зоны инфаркта миокарда, а также тормозит реваскуляризацию ишемизированной конечности у этих мышей [25]. Связывание адипонектина с Т-кадгерином в миокарде приводит к активации АМРК, что обеспечивает регенерацию. В свете перечисленных данных можно предположить, что Т-кадгерин представляет собой рецептор адипо-нектина или является ко-рецептором AdipoR1 или AdipoR2, опосредующим рекрутирование адипо-нектина в органы и ткани и обеспечивающим его протективное действие. При связывании с адипо-нектином Т-кадгерин неизвестным образом участвует в передаче сигнала с поверхности мембраны и регулирует экспрессию гена адипонектина.
В литературе накоплены данные о том, что Т-кадгерин опосредует эффекты адипонектина на рост сосудов, причем эти эффекты могут быть различны при физиологическом ангиогенезе или регенерации и при неоваскуляризации опухолей. Анализ параметров кровотока в ишемизированной конечности у мышей показал, что взаимодействие Т-кадгерина с адипонектином необходимо для полноценной реваскуляризации, так как у мышей, нокаутных по адипонектину или по Т-кадгерину, эффективного восстановления кровотока практически не происходит [25]. Отрицательное влияние T-кадгерина на начальные этапы ангиогенеза, обусловленное гомофильным взаимодействием между молекулами Т-кадгерина на эндотелиальных клетках сосудов и на клетках стромы, было продемонстрировано на физиологической модели васкуляризации Матригеля у мышей in vivo [26]. Аналогичные данные были получены и на модели неоваскуляризации первичного опухолевого узла меланомы у мышей: гиперэкспрессия Т-кадгерина в клетках меланомы подавляла врастание Т-кадгерин-экспрессирующих сосудов в первичный опухолевый узел, в то же время такие клетки приобретали способность активировать окружающую строму и имели повышенный метастатический и инвазивный потенциал [27].
В эндотелиальных клетках, выстилающих синусоидные капилляры печени, в норме и при таких патологиях, как хронический гепатит и цирроз печени, экспрессия Т-кадгерина практически отсутствует, однако в гепатоцеллюлярной карциноме человека Т-кадгерин экспрессируется в большом количестве в сосудах, прорастающих в опухолевые узлы. Экспрессия Т-кадгерина увеличивается по мере прогрессии заболевания и коррелирует со стадией гепатоцеллюлярной карциномы, что позволило авторам высказать предположение о стимулирующей роли Т-кадгерина при опухолевом неоангиогенезе.
На модели рака молочной железы у мышей, экспрессирующих MMTV-PyV-mT (Mouse Mammary Tumor Virus Promoter) и нокаутных по Т-кадгерину, было обнаружено, что в отсутствие Т-кадгерина неоваскуляризация опухолевых узлов существенно замедлена, что сопровождается гипоксией и увеличением количества легочных метастазов. У этих мышей отмечается снижение содержания адипонектина, связанного с сосудистой сетью, прорастающей в опухоль, и увеличение его содержания в плазме крови [28]. Авторы сделали вывод о том, что Т-кадгерин стимулирует опухолевый неоангиогенез in vivo и обеспечивает связывание адипонектина с опухолевыми сосудами, что, возможно, опосредует антиканцерогенные эффекты адипонектина.
Данные о роли Т-кадгерина в процессах формирования и прогрессирования злокачественных новообразований в литературе противоречивы [29]. Достаточно давно было высказано мнение о том, что Т-кадгерин является супрессором опухолевого роста. В ряде опухолей потеря экспрессии Т-кадгерина, связанная с хромосомными перестройками или гиперметилированием промотора гена Т-кадгерина, ассоциирована с опухолевым ростом и метастазированием [30]. Снижение экспрессии Т-кадгерина коррелирует с развитием рака молочной железы, рака легких, желчного пузыря, базальноклеточного, метатипического и плоскоклеточного рака кожи, меланомы [31], рака головы и шеи и ранних стадий рака поджелудочной железы, лейкемии, лимфом и ретинобластомы [32]. В то же время снижение экспрессии Т-кадгерина при раке яичников и эндометрия, а также при остеосаркоме, наоборот, ассоциировано с лучшей выживаемостью. На ранних стадиях рака простаты экспрессия белка Т-кадгерина значительно повышается, что существенно изменяется на более поздних этапах [32]. В нейробластоме экспрессия Т-кадгерина способствует подавлению опухолевого роста за счет ингибирования EGF-опосредованного сигнального пути и задержки пролиферации опухолевых клеток, в то время как в астроцитах повышение экспрессии Т-кадгерина связано со злокачественной трансформацией, возникновением глиом и глиобластом [32].
В исследовании с использованием метаанализа выявлена значимая ассоциация между степенью метилирования промотора Т-кадгерина и риском возникновения КРР. Авторами Т-кадгерин был назван значимым маркером ранней детекции колоректального рака [33].
Следует отметить, что лишь в отдельных работах, посвященных исследованию роли Т-кадгерина в опухолевой прогрессии, авторы анализировали уровень содержания адипонектина в плазме крови пациентов с опухолями или влияние адипонек-тина на фенотип линейных опухолевых клеток. Так, анализ содержания HMW адипонектина, AdipoR2 (но не AdipoR1) и Т-кадгерина показал значительное снижение содержания этих белков в образцах плазмы крови больных колоректальным раком по сравнению с условно здоровыми людьми, причем степень снижения адипонектина коррелировала со стадией заболевания [34]. На линейных клетках колоректального рака CaCo-2 и HCT116 было продемонстрировано, что ади-понектин подавляет выживаемость и миграцию клеток, а также индуцирует окислительный стресс. Кроме того, адипонектин вызывает изменение содержания мРНК провоспалительных (IL-6 и IL-8) и противовоспалительных (IL-10) цитокинов [35]. В другом исследовании на HCT116 клетках было обнаружено, что адипонектин вызывает снижение мРНК COX-2, но увеличивает содержание мРНК Т-кадгерина, что в сочетании с данными пациентов с колоректальным раком позволило сделать вывод об антиканцерогенной активности адипонектина. Таким образом, механизмы участия Т-кадгерина в опухолевой прогрессии не ясны, но, возможно, изменение экспрессии Т-кадгерина, необходимого для фиксации адипонектина в органах и тканях, может служить ранним маркером опухолевой трансформации клеток разного типа.
Резистин
Резистин является членом семейства резистиноподобных молекул, циркулирующим в плазме крови в виде димерного белка. Помимо клеток жировой ткани, резистин секретируется моно-нуклеарными клетками периферической крови, макрофагами, клетками костного мозга и поджелудочной железы. Физиологические эффекты резистина обусловлены его активностью, ассоциированной с развитием диабета и воспаления. В настоящее время идентифицированы четыре рецептора резистина: Толл-подобный рецептор 4 (TLR4), белок, ассоциированный с аденилци-клазой-1 (CAP1), изоформа декорина (ΔDCN) и трансмембранная тирозиновая киназа ROR1; специфичность сигнальных эффектов резистина зависит от того, с каким рецептором он взаимодействует. Провоспалительные эффекты резистина были впервые показаны при его взаимодействии с TLR4 рецептором на линии эпителиальных клеток почки эмбриона человека [36]. Связывание резистина с CAP1 активирует сигнализацию с участием фактора транскрипции NF-κB с последующей индукцией экспрессии провоспалитель-ных цитокинов IL-6, TNF-α и IL-1B, и ряда генов, участвующих в развитии инсулинорезистентности, воспаления и в апоптозе. Резистин-опосредованная активация другого фактора транскрипции, STAT3, способствует росту, агрессивному фенотипу и селекции раковых стволовых клеток при раке молочной железы [37]. Недавно проведенный метаанализ показал взаимосвязь между высоким уровнем продукции резистина и риском развития опухолевых заболеваний (рак молочной железы, эндометрия и колоректальный рак) у пациентов, страдающих ожирением. В то же время авторы отметили, что высокий уровень резистина не является предиктором возникновения опухоли, ассоциированной с ожирением.
В одном из исследований показано, что у больных РМЖ (в постменопаузе и пременопаузе) сывороточная концентрация резистина положительно коррелирует с систолическим давлением, уровнем триглицеридов, холестерина и отрицательно коррелирует с ЛПВП, в то время как у здоровых людей корреляции сывороточных концентраций адипо-кинов с основными метаболическими и антропометрическими параметрами отсутствуют. Кроме того, в этом же исследовании показана ассоциация сывороточного уровня резистина с размером и стадией опухоли, а также метастазированием в постменопаузе [38]. При обследовании пациентов с метастатическим колоректальным раком отмечено повышение концентрации резистина в сыворотке крови. В другом клиническом исследовании сообщалось, что сывороточный уровень резистина был повышен у больных КРР с метастазами в отличие от пациентов с ожирением, но без опухолевых заболеваний в анамнезе [39].
Опубликованные данные о взаимосвязи резистина с возникновением опухолевых заболеваний и опухолевой прогрессией получены в основном в результате обследования пациентов с избыточной массой тела, однако появляются данные о возможном участии резистина в развитии раковой кахексии. Анализ содержания резистина у больных немелкоклеточным раком легкого и гастроэзофагеальным раком в стадии кахексии показал ассоциацию между высоким уровнем резистина и возникновением метастазов по сравнению с контрольной группой больных, у которых регистрировали нормальный уровень резистина [40].
Висфатин
Роль висфатина в развитии раковой кахексии все еще остается до конца непонятой. В белой жировой ткани онкобольных с кахексией, а также у животных на экспериментальных моделях с верифицированной кахексией были проанализированы вариации ко-экспрессии висфатина с белками, связанными с липолизом. Уровень экспрессии мРНК висфатина был значительно выше у пациентов с кахексией, а также у животных на промежуточной стадии кахексии; в то же время, несмотря на увеличение мРНК, концентрация висфатина на уровне белка оставалась неизменной. При этом, в отличие от экспериментов на животных, у пациентов с терминальной стадией кахексии экспрессия висфатина как на уровне экспрессии мРНК, так и на уровне белка в циркулирующей крови была значительно снижена [43]. Аналогичные изменения отмечены и в отношении снижения содержания адипонектина, что, возможно, отражает метаболические изменения и деградацию жировой ткани при прогрессировании кахексии.
Апелин
Апелин – секретируемый адипокин, свойства которого подобны лиганду рецептора ангиотензина-I(II). Помимо жировой ткани апелин экспрессируется клетками других тканей, включая клетки ЦНС, легких, печени и сердца [44]. Его роль в качестве адипокина остается до конца непонятной. По своим эффектам апелин во многом похож на инсулин; он обладает провоспалительными свойствами и регулирует артериальное давление. В литературе имеются данные, свидетельствующие о том, что апелин способствует развитию заболеваний, связанных с ожирением, а также возникновению и прогрессированию опухолевых заболеваний, где его функция сводится к стимуляции ангиогенеза. На экспериментальных моделях показано, что высокая экспрессия апелина при ожирении способствует прогрессии РМЖ, а использование антагониста его рецептора при ожирении замедляет опухолевый рост [45].
При оценке взаимосвязи раковой кахексии и апелина недавно было показано, что значительно более высокий уровень апелина наблюдается в сыворотке крови и тканях у пациентов с гастроэзофагеальным раком в стадии кахексии по сравнению с контрольной группой. Однако корреляции между уровнем апелина и состоянием кахексии, а также с какими-либо клинико-патологическими параметрами не обнаружено [46].
Адипсин
Адипсин (фактор комплемента D, CFD) является одним из первых описанных адипокинов, который секретируется в значительных количествах адипоцитами. Ген адипсина является прямой мишенью фактора транскрипции PPARγ (рецептор, активируемый пролифераторами пероксисом, гамма) [47]. Адипсин контролирует альтернативный путь активации комплемента, стимулируя выработку С3а (активной формы компонента комплемента 3), и опосредованно участвует в таких процессах, как иммунный ответ, миграция клеток, дифференцировка адипоцитов и хоуминг гемопоэтических стволовых клеток, а также влияет на инсулиноре-зистентность [47].
Роль адипсина в развитии и прогрессии рака мало исследована. В одной из немногочисленных публикаций говорится о влиянии адипсина, секретируемого зрелыми адипоцитами, на активацию процесса аутофагии в клетках меланомы в ответ на химиотерапию [48]. В 2019 г. проведено исследование, в котором попытались выявить возможную взаимосвязь между адипсином, ожирением и онкогенезом. Обнаружено, что секретируемый жировой тканью молочной железы адипсин усиливает пролиферацию опухолевых стволовых клеток (ОСК) при раке молочной железы, причем, у пациенток с ожирением секреция адипсина была выражена сильнее. Дальнейшие исследования in vitro обнаружили способность адипсина стимулировать формирование сфероидов ОСК, а ингибирование адипсин-зависимой сигнализации подавляло как эту способность, так и экспрессию маркеров ство-ловости в ОСК [47]. Эти данные впервые позволили сформулировать гипотезу о том, что адипсин, секретируемый жировой тканью молочной железы, может быть важным компонентом ниши ОСК при раке молочной железы, в связи с чем актуальными являются исследования, направленные на выявление взаимосвязи между экспрессией адипсина и повышенным риском развития рака молочной железы у пациентов с ожирением, а также на изучение конкретных механизмов, обусловливающих действие адипсина при этом заболевании.
Лептин
Пептидный гормон лептин имеет молекулярную массу 16 кДа, секретируется адипоцитами и играет ключевую роль в регулировании энергетического баланса, снижая аппетит и увеличивая интенсивность метаболизма. Так как уровень лептина у людей с ожирением повышен, существует предположение, что при ожирении формируется резистентность клеток к лептину. Некоторые опухолевые заболевания, а именно рак прямой кишки, молочной железы и эндометрия, характеризуются повышенной экспрессией рецептора лептина ObR. Связываясь со своим рецептором ObR на клеточной поверхности, лептин активирует транскрипционный фактор STAT3, что приводит к нарушению регуляции апоптоза, как было показано на линейных клетках рака молочной железы, рака ободочной кишки и низкодифференцированных карцином яичника [49].
Лептин оказывает митогенное и антиапоптоти-ческое действие на линейные клетки таких опухолевых линий, как клетки рака молочной железы, пищевода, толстой кишки и простаты, однако он ингибирует пролиферацию клеток рака поджелудочной железы [50]. На линейных клетках рака эндометрия и колоректального рака были получены данные о лептин-индуцированной пролиферации, инвазии и миграции клеток за счет сигнализации с участием PI3K/Akt/mTOR сигнальных путей, которые активируются при связывании фактора роста IGF-1 со своим рецептором, что свидетельствует о взаимосвязи метаболических нарушений и процессов канцерогенеза. Подавление активности MAPK и PI3K блокирует митогенные эффекты лептина на линейных опухолевых клетках [50]. Следует отметить, что значительная часть этих неблагоприятных эффектов лептина блокируется адипонектином как in vitro , так и in vivo [51].
Половые гормоны
Влияние ожирения на синтез и биодоступность эндогенных половых гормонов имеет важное значение для понимания механизмов возникновения рака молочной железы и эндометрия у женщин с избыточной массой тела. При ожирении увеличивается превращение андрогенных предшественников в эстрадиол ароматазой в клетках жировой ткани, что ведет к повышению уровня эстрадиола в сыворотке крови, который оказывается не сбалансирован уровнем прогестерона. Повышенный уровень инсулина в сыворотке крови в результате дисфункции жировой ткани может приводить как к увеличению синтеза андрогенов в яичниках, так и к снижению синтеза глобулина в печени, который связывает и инактивирует половые гормоны. Последние данные о повышении в плазме крови концентрации эстрадиола и тестостерона и снижении концентрации глобулина, связывающего половые гормоны, у женщин в постменопаузе с ожирением подтверждают взаимозависимость метаболизма половых гормонов и ожирения [55], что необходимо учитывать при разработке мер профилактики рака, ассоциированного с ожирением.
Взаимосвязь эндогенных половых гормонов и опухолевой прогрессии при раке молочной железы и раке эндометрия достоверно установлена. Проспективные исследования демонстрируют, что уровень эндогенных половых гормонов тесно связан с риском их развития в постменопаузе [56]. Предполагается, что пролиферативное действие эстрогена на эпителиальную ткань молочной железы и эндометрия является основным механизмом запуска канцерогенеза.
Система активаторов плазминогена: урокиназа uPA, ее рецептор uPAR и ингибитор активаторов плазминогена PAI-1 Система активатора плазминогена состоит из урокиназы uPA, её рецептора uPAR и двух специфических ингибиторов PAI-1 и PAI-2. uPA превращает неактивный плазминоген в плазмин, который в кровеносном русле запускает процесс фибринолиза, а в ткани – ремоделирование базальной мембраны и внеклеточного матрикса (ВКМ). Данные последних лет указывают на то, что плазминоген и урокиназная система играют важную роль в целом ряде процессов, связанных с опухолевой прогрессией, а именно стимулируют опухолевый неоангиогенез, а также интравазацию, инвазию и метастазирование опухолевых клеток и окружающей стромы. Повышенная экспрессия uPA, uPAR и PAI-1 обнаружена почти во всех типах опухолей; более того, в ряде случаев наблюдается высокая положительная корреляция между уровнем экспрессии этих белков в ткани опухоли и плохим прогнозом как в отношении риска появления метастаз, так и химиорезистентности. При исследовании роли урокиназной системы в опухолевой прогрессии было обнаружено, что экспрессия uPA и uPAR часто оказывается повышена и свидетельствует о неблагоприятном течении заболевания [57]. Для клеток меланомы человека in vitro было продемонстрировано, что сигнальные эффекты uPAR опосредованы латеральным взаимодействием uPAR с α5β1-интегринами и через них с рецептором эпидермального фактора роста (ЕGFR), высокая экспрессия и активность которого отмечены в различных типах опухолей помимо меланомы [58]. Результатом формирования комплекса uPAR с α5β1-интегринами и ЕGFR является активация PI3K-mTOR-HIFα сигнального пути, что повышает способность опухолевых клеток к инвазии и увеличивает интенсивность гликолиза. Таким образом, uPA/uPAR система представляет собой ключевую точку регуляции как фенотипических свойств (инвазия и метастатический потенциал) клеток меланомы, так и их гликолитического метаболизма. Это позволяет рассматривать uPA/uPAR в качестве перспективной мишени при разработке адъювантных или самостоятельных подходов к лечению меланомы [58].
PAI-1 является ингибитором сериновых протеаз, его экспрессия обнаруживается в адипоцитах, эндотелиальных и стромальных клетках жировой ткани. PAI-1 не только продуцируется адипоцитами жировой ткани, но и влияет на их дифференцировку и инсулин-зависимую сигнализацию. Несмотря на то, что PAI-1 ингибирует урокиназу, которая стимулирует деградацию ВКМ и инвазию опухолевых клеток, сам PAI-1 оказывает стимулирующее влияние на их рост, инвазию и метастазирование. В основе этих эффектов лежат взаимодействие PAI-1 с белками ВКМ и рецепторами ВКМ и их активация. Увеличение экспрессии PAI-1 было обнаружено при многих типах рака, ассоциированных с ожирением, и коррелирует с прогрессией рака молочной железы, эндометрия, КРР, рака щитовидной железы, почки и простаты. Помимо аутокринной продукции PAI-1 опухолевыми клетками, системный уровень PAI-1, вероятно, также имеет важное значение для стимуляции опухолевого роста [59]. У мышей линии Min, дефектных по гену белка Apc (Adenomatous polyposis coli), терапия ингибитором PAI-1 снижала образование полипов в кишечнике [54]. Недавно появились данные о том, что активация экспрессии PAI-1, характерная для MC, ассоциирована с течением рака молочной железы у женщин по более агрессивному сценарию. Предположительно, стимулирующая роль PAI-1 в канцерогенезе и опухолевом росте обусловлена именно его способностью усиливать миграцию клеток и опухолевый неоангиогенез.
Одна из характерных черт клеток опухоли – способность продуцировать факторы роста и протеолитические ферменты, обеспечивающие деградацию и ремоделирование окружающего матрикса, инвазию клеток в окружающие ткани, неоангиогенез, рост опухолевых масс и метастазирование. Увеличение экспрессии и/или активности урокиназы, повышение содержания рецептора урокиназы в ткани опухоли или окружающей строме коррелируют с негативным клиническим прогнозом [60]. Экспрессия uPAR регулируется некоторыми стероидными гормонами и факторами роста. Показано значимое влияние uPA и uPAR на инвазию и метастазирование гормонозависимых видов рака: прежде всего рака простаты и РМЖ. 8-мерный пептид А6, используемый как антагонист uPA и обладающий антиангиогенным и проапоп-тотическим эффектами, блокирует опухолевую прогрессию гормон-чувствительного рака молочной железы в экспериментах на крысах in vivo и линейных раковых клетках in vitro. In vivo введение А6 пептида усиливает противоопухолевые эффекты гормональной терапии при использовании его в комбинации с тамоксифеном (антиэстрогеном) на сингенной модели рака молочной железы у крыс. Эти исследования показывают, что применение такого рода агентов в сочетании с гормональной терапией может повышать эффективность лечения за счёт ингибирования ангиогенеза и индукции гибели опухолевых клеток [61].
Воспаление
Злокачественные новообразования часто сопровождаются воспалительными тканевыми реакциями. Воспалительное опухолевое микроокружение продуцирует большое количество провоспалитель-ных факторов и цитокинов. Роль этих факторов и механизмы их действия при канцерогенезе до конца не ясны, однако в разных исследованиях было продемонстрировано их важное прогностическое значение [62]. Ожирение можно рассматривать как вариант слабовыраженного системного воспаления. Жировая ткань является не только производителем, но и тканевым депо провоспалительных цитокинов: TNF-α, IL-1, IL-6, MCP-1 (моноцитарный хемотаксический фактор 1) и др. Можно предположить, что длительная циркуляция этих цитокинов в крови может как стимулировать опухолевую трансформацию клеток (так как эти цитокины обладают митогенным эффектом на клетки), так и поддерживать уже сформированное опухолью воспалительное микроокружение [63, 64]. Так, прямое участие в запуске канцерогенеза было показано на примере TNF-α – одного из наиболее известных факторов воспаления, который участвует не только в злокачественной трансформации на ранних, но и на последующих этапах опухолевой прогрессии. Сигнализация TNF-α через рецептор TNF-R1 приводит к активации NF-κB, что, в свою очередь, активирует белки c-FLIP и cIAP1, которые являются факторами, подавляющими апоптоз и способствующими выживанию клеток. Кроме того, некоторые опухолевые клетки сами продуцируют TNF-α. Обнаружено, что TNF-α, который продуцируется клетками рака яичников, стимулирует ряд факторов, включая фактор роста сосудов VEGF и хемокины CXCR4 и CXCL12, которые способствуют прогрессированию заболевания. Точные сигнальные механизмы действия TNF-α, повышение которого характерно как для ожирения, так и для опухолевого роста, до конца не ясны, но известно, что повышенный уровень TNF-α в сыворотке крови при опухолевом заболевании коррелирует с высоким риском летальности и, в меньшей степени, с неблагоприятным прогнозом [65]. Показано, что повышение системного уровня TNF-α играет значимую роль на самых ранних этапах некоторых опухолевых заболеваний. Так, высокий уровень TNF-α достоверно ассоциирован с повышенным риском развития колоректальной аденомы [66].
В норме провоспалительный цитокин IL-6 играет важную роль при остром воспалительном ответе и участвует в созревании В-клеток. Однако есть данные, указывающие на его тесную взаимосвязь с хроническим воспалением и раком. Уровень IL-6 повышен в плазме крови при ожирении, и, подобно TNF-α, высокий уровень IL-6 коррелирует с общей смертностью от рака и повышенным риском возникновения предраковых состояний. Увеличение активности промотера IL-6 отмечается при некоторых типах гемобластозов [66]. Влияние IL-6 на пролиферацию и выживаемость клеток опосредовано через активацию JAK-сигнального пути (янус-киназа JAK) и белка STAT3 семейства транскрипционных факторов. Нами показано, что рецептор урокиназы uPAR может регулировать экспрессию IL-6 в клетках нейробластомы, причем повышение его экспрессии ассоциировано с активацией программы эпителиально-мезенхимального перехода [67]. Кроме того, высокая экспрессия урокиназного рецептора в опухолях обнаруживается не только в виде мембранно-связанного белка, но и в виде растворимой формы (suPAR, soluble uPAR). Растворимый uPAR является мощным хемоаттрактантом для клеток иммунной системы, в частности нейтрофилов, и способен привлекать их в очаг воспаления через активацию хемокиновых рецепторов fMLP пептида [68]. Помимо функции uPAR, которая связана с его взаимодействием с урокиназой, именно способность uPAR поддерживать воспаление объясняет неудачи, связанные с использованием блокирования урокиназы при лечении новообразований, и делает необходимым дальнейшее изучение свойств урокиназного рецептора в клетках опухоли и окружающей её провос-палительной стромы.
Воспалительная реакция, сопровождающая ожирение, включает и другие компоненты, способствующие опухолевой прогрессии. Среди них следует отметить матриксные металлопротеина- зы (ММР), активность которых регулируется в том числе и урокиназой и которые обеспечивают инвазию опухолевых клеток и метастазирование. Индукция мРНК некоторых ММР при ожирении, а также описанное влияние ММР на дифференцировку преадипоцитов дополнительно свидетельствуют о вероятной взаимосвязи ожирения с риском возникновения опухоли [69]. Окислительный стресс, как составная часть хронического воспаления, может также создавать микроокружение, благоприятное для возникновения и прогрессирования опухолевых заболеваний при ожирении.
Заключение
Детали молекулярных и клеточных механизмов влияния избыточного веса на возникновение и прогрессирование опухолевых заболеваний мало изучены. Мало известно о взаимосвязи нарушения функции жировой ткани с инсулинорезистентно-стью, изменением содержания половых гормонов в плазме крови и клиническим прогнозом при
Список литературы Влияние ожирения на развитие и прогрессию злокачественных новообразований: обзор современных данных и новых терапевтических мишеней
- Yunusova N.V., Kondakova I.V., Kolomiets L.A., Afanas'ev S.G., Kishkina A.Y., Spirina L.V. The role of metabolic syndrome variant in the malignant tumors progression. Diabetes Metab Syndr. 2018; 12(5): 807-812. https://doi.org/10.1016/j.dsx.2018.04.028.
- Harvey I., Boudreau A., Stephens J.M. Adipose tissue in health and disease. Open Biol. 2020 Dec; 10(12): 200291. https://doi.org/10.1098/rsob.200291.
- De Pergola G., Silvestris F. Obesity as a major risk factor for cancer. J Obes. 2013; 2013: 291546. https://doi.org/10.1155/2013/291546.
- Amin M.N., Hussain M.S., Sarwar M.S., Rahman Moghal M.M., Das A., Hossain M.Z., Chowdhury J.A., Millat M.S., Islam M.S. How the association between obesity and inflammation may lead to insulin resistance and cancer. Diabetes Metab Syndr. 2019; 13(2): 1213-24. https://doi.org/10.1016/j.dsx.2019.01.041.
- Ziemke F., Mantzoros C.S.Adiponectin in insulin resistance: lessons from translational research. Am J Clin Nutr. 2010 Jan; 91(1): 258S-261S. https://doi.org/10.3945/ajcn.2009.28449C.
- Porporato P.E. Understanding cachexia as a cancer metabolism syndrome. Oncogenesis. 2016 Feb 22; 5(2): e200. https://doi.org/10.1038/oncsis.2016.3.
- Yoon Y.S., Kwon A.R., Lee Y.K., Oh S.W. Circulating adipokines and risk of obesity related cancers: A systematic review and meta-analysis. Obes Res Clin Pract. 2019 Jul-Aug; 13(4): 329-339. https://doi.org/10.1016/j.orcp.2019.03.006.
- Rubio-Jurado B., Balderas-Peña L.M., García-Luna E.E., ZavalaCerna M.G., Riebeling-Navarro C., Reyes P.A., Nava-Zavala A.H. Obesity, Thrombotic Risk, and Inflammation in Cancer. Adv Clin Chem. 2018; 85: 71-89. https://doi.org/10.1016/bs.acc.2018.02.006.
- Larsson S.C., Rutegård J., Bergkvist L., Wolk A. Physical activity, obesity, and risk of colon and rectal cancer in a cohort of Swedish men. Eur J Cancer. 2006 Oct; 42(15): 2590-7. https://doi.org/10.1016/j.ejca.2006.04.015.
- Calle E.E., Rodriguez C., Walker-Thurmond K., Thun M.J. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med. 2003; 348(17): 1625-38. https://doi.org/10.1056/NEJMoa021423.
- Renehan A.G., Tyson M., Egger M., Heller R.F., Zwahlen M. Bodymass index and incidence of cancer: a systematic review and meta-analysis of prospective observational studies. Lancet. 2008 Feb 16; 371(9612): 569-78. https://doi.org/10.1016/S0140-6736(08)60269-X.
- Quail D.F., Dannenberg A.J. The obese adipose tissue microenvironment in cancer development and progression. Nat Rev Endocrinol. 2019 Mar; 15(3): 139-154. https://doi.org/10.1038/s41574-018-0126-x.
- Bergan-Roller H.E., Sheridan M.A. The growth hormone signaling system: Insights into coordinating the anabolic and catabolic actions of growth hormone. Gen Comp Endocrinol. 2018; 258: 119-33. https://doi.org/10.1016/j.ygcen.2017.07.028.
- Bauer T.W., Liu W., Fan F., Camp E.R., Yang A., Somcio R.J., Bucana C.D., Callahan J., Parry G.C., Evans D.B., Boyd D.D., Mazar A.P., Ellis L.M. Targeting of urokinase plasminogen activator receptor in human pancreatic carcinoma cells inhibits c-Met- and insulin-like growth factor-I receptor-mediated migration and invasion and orthotopic tumor growth in mice. Cancer Res. 2005 Sep 1; 65(17): 7775-81. https://doi.org/10.1158/0008-5472.CAN-05-0946.
- Tsujimoto T., Kajio H., Sugiyama T. Association between hyperinsulinemia and increased risk of cancer death in nonobese and obese people: A population-based observational study. Int J Cancer. 2017 Jul 1; 141(1): 102-111. https://doi.org/10.1002/ijc.30729.
- Ahmed H.H., Hameed E.R.A., Shalby A.B. E.-N.H. Potent role of lipocalin in childhood obesity. World J Med Sci. 2012; 7(2): 100-4.
- Di Zazzo E., Polito R., Bartollino S., Nigro E., Porcile C., Bianco A., Daniele A., Moncharmont B. Adiponectin as Link Factor between Adipose Tissue and Cancer. Int J Mol Sci. 2019 Feb 15; 20(4): 839. https://doi.org/10.3390/ijms20040839.
- Karnati H.K., Panigrahi M.K., Li Y, Tweedie D., Greig N.H. Adiponectin as a Potential Therapeutic Target for Prostate Cancer. Curr Pharm Des. 2017; 23(28): 4170-4179. https://doi.org/10.2174/1381612823666170208123553.
- Nunez N.P., Oh W.J., Rozenberg J., Perella C., Anver M., Barrett J.C., Perkins S.N., Berrigan D., Moitra J., Varticovski L., Hursting S.D., Vinson C. Accelerated tumor formation in a fatless mouse with type 2 diabetes and inflammation. Cancer Res. 2006 May 15; 66(10): 5469-76. https://doi.org/10.1158/0008-5472.CAN-05-4102.
- Batista M.L.Jr., Olivan M., Alcantara P.S., Sandoval R., Peres S.B., Neves R.X., Silverio R., Maximiano L.F., Otoch J.P., Seelaender M. Adipose tissue-derived factors as potential biomarkers in cachectic cancer patients. Cytokine. 2013 Feb; 61(2): 532-9. https://doi.org/10.1016/j.cyto.2012.10.023.
- Kyriakakis E., Frismantiene A., Dasen B., Pfaff D., Rivero O., Lesch K.P., Erne P., Resink T.J., Philippova M. T-cadherin promotes autophagy and survival in vascular smooth muscle cells through MEK1/2/Erk1/2 axis activation. Cell Signal. 2017 Jul; 35: 163-175. https://doi.org/10.1016/j.cellsig.2017.04.004.
- Rubina K., Talovskaya E., Cherenkov V., Ivanov D., Stambolsky D., Storozhevykh T., Pinelis V., Shevelev A., Parfyonova Y., Resink T., Erne P., Tkachuk V. LDL induces intracellular signalling and cell migration via atypical LDL-binding protein T-cadherin. Mol Cell Biochem. 2005 May; 273(1-2): 33-41. https://doi.org/10.1007/s11010-005-0250-5.
- Balatskaya M., Sharonov G., Baglay A., Balatskiy A. Tkachuk V. One receptor, two ligands, different responses: T-cadherin as a receptor for low density lipoprotein and adiponectin. FEBS J. 2017; 284: 102-403. https://doi.org/10.1111/febs.14174.
- Shehzad A., Iqbal W., Shehzad O., Lee Y.S. Adiponectin: Regulation of its production and its role in human diseases. Hormones. 2012 Jan; 11(1): 8-20. https://doi.org/10.1007/BF03401534.
- Parker-Duffen J.L., Nakamura K., Silver M., Kikuchi R., Tigges U., Yoshida S., Denzel M.S., Ranscht B., Walsh K. T-cadherin is essential for adiponectin-mediated revascularization. J Biol Chem. 2013 Aug 23; 288(34): 24886-97. https://doi.org/10.1074/jbc.M113.454835.
- Rubina K., Kalinina N., Potekhina A., Efimenko A., Semina E., Poliakov A., Wilkinson D.G., Parfyonova Y., Tkachuk V. T-cadherin sup presses angiogenesis in vivo by inhibiting migration of endothelial cells. Angiogenesis. 2007; 10(3): 183-95. https://doi.org/10.1007/s10456-007-9072-2.
- Rubina K.A., Surkova E.I., Semina E.V., Sysoeva V.Y., Kalinina N.I., Poliakov A.A., Treshalina H.M., Tkachuk V.A. T-Cadherin Expression in Melanoma Cells Stimulates Stromal Cell Recruitment and Invasion by Regulating the Expression of Chemokines, Integrins and Adhesion Molecules. Cancers (Basel). 2015 Jul 21; 7(3): 1349-70. https://doi.org/10.3390/cancers7030840.
- Hebbard L.W., Garlatti M., Young L.J., Cardiff R.D., Oshima R.G. Ranscht B. T-cadherin supports angiogenesis and adiponectin association with the vasculature in a mouse mammary tumor model. Cancer Res. 2008 Mar 1; 68(5): 1407-16. https://doi.org/10.1158/0008-5472.CAN-07-2953.
- Andreeva A.V., Kutuzov M.A. Cadherin 13 in cancer. Genes Chromosomes Cancer. 2010 Sep; 49(9): 775-90. https://doi.org/10.1002/gcc.20787.
- Rubina K.A., Sysoeva V.Y., Zagorujko E.I., Tsokolaeva Z.I., Kurdina M.I., Parfyonova Y.V., Tkachuk V.A. Increased expression of uPA, uPAR, and PAI-1 in psoriatic skin and in basal cell carcinomas. Arch Dermatol Res. 2017 Aug; 309(6): 433-442. https://doi.org/10.1007/s00403-017-1738-z.
- Rubina K.A., Sysoeva V.Yu., Semina E.V., Yurlova E.I., Molochkov V.A., Khlebnikova A.N., Sedova T.G. Osobennosti ekspressii T-kadgerina v keratinotsitakh i sosudakh epitelial'nykh opukholei kozhi. Rossiiskii zhurnal kozhnykh i venericheskikh boleznei. 2013; 2013(1): 9-14.
- McWilliam J., editor. Cadherins: Types, Structure and Functions. NY (USA): Nova Science Publishers; 2020. 171 p.
- Duan B.S., Xie L.F., Wang Y. Aberrant Methylation of T-cadherin Can Be a Diagnostic Biomarker for Colorectal Cancer. Cancer Genomics Proteomics. 2017 Jul-Aug; 14(4): 277-284. https://doi.org/10.21873/cgp.20038.
- Polito R., Nigro E., Fei L., DE Magistris L., Monaco M.L., D'Amico R., Naviglio S., Signoriello G., Daniele A. Adiponectin Is Inversely Associated With Tumour Grade in Colorectal Cancer Patients. Anticancer Res. 2020 Jul; 40(7): 3751-3757. https://doi.org/10.21873/anticanres.14364.
- Nigro E., Schettino P., Polito R., Scudiero O., Monaco M.L., De Palma G.D., Daniele A. Adiponectin and colon cancer: evidence for inhibitory effects on viability and migration of human colorectal cell lines. Mol Cell Biochem. 2018 Nov; 448(1-2): 125-135. https://doi.org/10.1007/s11010-018-3319-7.
- Tarkowski A., Bjersing J., Shestakov A., Bokarewa M.I. Resistin competes with lipopolysaccharide for binding to toll-like receptor 4. J Cell Mol Med. 2009 Sep; 14(6b): 1419-31. https://doi.org/10.1111/j.1582-4934.2009.00899.x.
- Deshmukh S.K., Srivastava S.K., Zubair H., Bhardwaj A., Tyagi N., Al-Ghadhban A., Singh A.P., Dyess D.L., Carter J.E., Singh S. Resistin potentiates chemoresistance and stemness of breast cancer cells: Implications for racially disparate therapeutic outcomes. Cancer Lett. 2017 Jun 28; 396: 21-29. https://doi.org/10.1016/j.canlet.2017.03.010.
- Assiri A.M., Kamel H.F., Hassanien M.F. Resistin, visfatin, adiponectin, and leptin: risk of breast cancer in pre- and postmenopausal saudi females and their possible diagnostic and predictive implications as novel biomarkers. Dis Markers. 2015; 2015: 253519. https://doi.org/10.1155/2015/253519.
- Nakajima T.E., Yamada Y., Hamano T., Furuta K., Matsuda T., Fujita S., Kato K., Hamaguchi T., Shimada Y. Adipocytokines as new promising markers of colorectal tumors: adiponectin for colorectal adenoma, and resistin and visfatin for colorectal cancer. Cancer Sci. 2010 May; 101(5): 1286-91. https://doi.org/10.1111/j.1349-7006.2010.01518.x.
- Demiray G., Değirmencioğlu S., Uğurlu E., Yaren A. Effects of Serum Leptin and Resistin Levels on Cancer Cachexia in Patients With Advanced-Stage Non-Small Cell Lung Cancer. Clin Med Insights Oncol. 2017 Feb 20; 11: 1179554917690144. https://doi.org/10.1177/1179554917690144.
- Yaku K., Okabe K., Hikosaka K., Nakagawa T. NAD Metabolism in Cancer Therapeutics. Front Oncol. 2018 Dec 12; 8: 622. https://doi.org/10.3389/fonc.2018.00622.
- Lin T.C. The role of visfatin in cancer proliferation, angiogenesis, metastasis, drug resistance and clinical prognosis. Cancer Manag Res. 2019 Apr 23; 11: 3481-3491. https://doi.org/10.2147/CMAR.S199597.
- Silvério R., Lira F.S., Oyama L.M., Oller do Nascimento C.M., Otoch J.P., Alcântara P.S.M., Batista M.L.Jr., Seelaender M. Lipases and lipid droplet-associated protein expression in subcutaneous white adipose tissue of cachectic patients with cancer. Lipids Health Dis. 2017 Aug; 16(1): 159. https://doi.org/10.1186/s12944-017-0547-x.
- Wysocka M.B., Pietraszek-Gremplewicz K., Nowak D. The Role of Apelin in Cardiovascular Diseases, Obesity and Cancer. Front Physiol. 2018 May 23; 9: 557. https://doi.org/10.3389/fphys.2018.00557.
- Gourgue F., Mignion L., Van Hul M., Dehaen N., Bastien E., Payen V., Leroy B., Joudiou N., Vertommen D., Bouzin C., Delzenne N., Gallez B., Feron O., Jordan B.F., Cani P.D. Obesity and triple-negativebreast-cancer: Is apelin a new key target? J Cell Mol Med. 2020; 24(17): 10233-44. https://doi.org/10.1111/jcmm.15639.
- Diakowska D., Markocka-Mączka K., Szelachowski P., Grabowski K. Serum levels of resistin, adiponectin, and apelin in gastroesophageal cancer patients. Dis Markers. 2014; 2014: 619649. https://doi.org/10.1155/2014/619649.
- Goto H., Shimono Y., Funakoshi Y., Imamura Y., Toyoda M., Kiyota N., Kono S., Takao S., Mukohara T., Minami H. Adipose-derived stem cells enhance human breast cancer growth and cancer stem cell-like properties through adipsin. Oncogene. 2019; 38(6): 767-779. https://doi.org/10.1038/s41388-018-0477-8.
- Liu Z., Xu J., He J., Liu H., Lin P., Wan X., Navone N.M, Tong Q., Kwak L.W., Orlowski R.Z., Yang J. Mature adipocytes in bone marrow protect myeloma cells against chemotherapy through autophagy activation. Oncotarget. 2015 Oct 27; 6(33): 34329-41. https://doi.org/10.18632/oncotarget.6020.
- Kato S., Abarzua-Catalan L., Trigo C., Delpiano A., Sanhueza C., García K., Ibañez C., Hormazábal K., Diaz D., Brañes J., Castellón E., Bravo E., Owen G., Cuello M.A. Leptin stimulates migration and invasion and maintains cancer stem-like properties in ovarian cancer cells: an explanation for poor outcomes in obese women. Oncotarget. 2015; 6(25): 21100-19. https://doi.org/10.18632/oncotarget.4228.
- Zhou W., Tian Y., Gong H., Guo S., Luo C. Oncogenic role and therapeutic target of leptin signaling in colorectal cancer. Expert Opin Ther Targets. 2014 Aug; 18(8): 961-71. https://doi.org/10.1517/14728222.2014.926889.
- Wu X., Yan Q., Zhang Z., Du G., Wan X. Acrp30 inhibits leptininduced metastasis by downregulating the JAK/STAT3 pathway via AMPK activation in aggressive SPEC-2 endometrial cancer cells. Oncol Rep. 2012 May; 27(5): 1488-96. https://doi.org/10.3892/or.2012.1670.
- Kerem M., Ferahkose Z., Yilmaz U.T., Pasaoglu H., Ofluoglu E., Bedirli A., Salman B., Sahin T.T., Akin M. Adipokines and ghrelin in gastric cancer cachexia. World J Gastroenterol. 2008; 14(23): 3633-41. https://doi.org/10.3748/wjg.14.3633.
- Huang Q., Fan Y.-Z., Ge B.-J., Zhu Q., Tu Z.-Y. Circulating Ghrelin in Patients with Gastric or Colorectal Cancer. Dig Dis Sci. 2007 Feb; 52(3): 803-9. https://doi.org/10.1007/s10620-006-9508-3.
- Singhal M., Vishnu M.V.R., Raju S.V., Upadhyay Y. Interrelationship between obesity and cancer (A Review). Acad J Cancer Res. 2013; 6(1): 13-20. https://doi.org/10.5829/idosi.ajcr.2013.6.1.739.
- Baglietto L., English D.R., Hopper J.L., MacInnis R.J., Morris H.A., Tilley W.D., Krishnan K., Giles G.G. Circulating steroid hormone concentrations in postmenopausal women in relation to body size and composition. Breast Cancer Res Treat. 2009 May; 115(1): 171-9. https://doi.org/10.1007/s10549-008-0069-3.
- Brown S.B., Hankinson S.E. Endogenous estrogens and the risk of breast, endometrial, and ovarian cancers. Steroids. 2015 Jul; 99(Pt A): 8-10. https://doi.org/10.1016/j.steroids.2014.12.013.
- Noh H., Hong S., Huang S. Role of urokinase receptor in tumor progression and development. Theranostics. 2013 Jun; 3(7): 487-95. https://doi.org/10.7150/thno.4218.
- Laurenzana A., Chillà A., Luciani C., Peppicelli S., Biagioni A., Bianchini F., Tenedini E., Torre E., Mocali A., Calorini L., Margheri F., Fibbi G., Del Rosso M. uPA/uPAR system activation drives a glycolytic phenotype in melanoma cells. Int J Cancer. 2017 Sep 15; 141(6): 1190-1200. https://doi.org/10.1002/ijc.30817.
- Kubala M.H., DeClerck Y.A. The plasminogen activator inhibitor-1 paradox in cancer: a mechanistic understanding. Cancer Metastasis Rev. 2019 Sep; 38(3): 483-492. https://doi.org/10.1007/s10555-019-09806-4.
- Mahmood N., Mihalcioiu C., Rabbani S.A. Multifaceted Role of the Urokinase-Type Plasminogen Activator (uPA) and Its Receptor (uPAR): Diagnostic, Prognostic, and Therapeutic Applications. Front Oncol. 2018 Feb 12; 8: 24. https://doi.org/10.3389/fonc.2018.00024.
- Guo Y., Mazar A.P., Lebrun J.J., Rabbani S.A. An antiangiogenic urokinase-derived peptide combined with tamoxifen decreases tumor growth and metastasis in a syngeneic model of breast cancer. Cancer Res. 2002 Aug 15; 62(16): 4678-84.
- Danilova N.V., Mikhailov I.A., Oleinikova N.A., Mal'kov P.G., Chaika A.V., Khomyakov V.M., Kakotkin V.V., Yudin M.Yu. Persistentsiya antigenov virusa Epshteina-Barr pri rake zheludka: kharakteristika vospalitel'nykh kletochnykh reaktsii v opukholi. Arkhiv patologii. 2021; 83(1): 18-24. https://doi.org/10.17116/patol20218301118.
- Deng T., Lyon C.J., Bergin S., Caligiuri M.A., Hsueh W.A. Obesity, Inflammation, and Cancer. Annu Rev Pathol. 2016 May 23; 11: 421-49. https://doi.org/10.1146/annurev-pathol-012615-044359.
- Kahn C.R., Wang G., Lee K.Y. Altered adipose tissue and adipocyte function in the pathogenesis of metabolic syndrome. J Clin Invest. 2019 Oct 1; 129(10): 3990-4000. https://doi.org/10.1172/JCI129187.
- Il'yasova D., Colbert L.H., Harris T.B., Newman A.B., Bauer D.C., Satterfield S., Kritchevsky S.B. Circulating levels of inflammatory markers and cancer risk in the health aging and body composition cohort. Cancer Epidemiol Biomarkers Prev. 2005 Oct; 14(10): 2413-8. https://doi.org/10.1158/1055-9965.EPI-05-0316.
- Zhang X., Liu S., Zhou Y. Circulating levels of C-reactive protein, interleukin-6 and tumor necrosis factor-α and risk of colorectal adenomas: a meta-analysis. Oncotarget. 2016; 7(39): 64371-79. https://doi.org/10.18632/oncotarget.11853.
- Semina E.V., Rubina K.A., Shmakova A.A., Rysenkova K.D., Klimovich P.S., Aleksanrushkina N.A., Sysoeva V.Y., Karagyaur M.N., Tkachuk V.A. Downregulation of uPAR promotes urokinase translocation into the nucleus and epithelial to mesenchymal transition in neuroblastoma. J Cell Physiol. 2020 Sep; 235(9): 6268-6286. https://doi.org/10.1002/jcp.29555.
- Pliyev B.K. Activated human neutrophils rapidly release the chemotactically active D2D3 form of the urokinase-type plasminogen activator receptor (uPAR/CD87). Mol Cell Biochem. 2009 Jan; 321(1-2): 111-22. https://doi.org/10.1007/s11010-008-9925-z.
- Cabral-Pacheco G.A., Garza-Veloz I., Castruita-De la Rosa C., Ramirez-Acuña J.M., Perez-Romero B.A., Guerrero-Rodriguez J.F., Martinez-Avila N., Martinez-Fierro M.L. The Roles of Matrix Metalloproteinases and Their Inhibitors in Human Diseases. Int J Mol Sci. 2020 Dec 20; 21(24): 9739. https://doi.org/10.3390/ijms21249739
