Врожденный буллезный эпидермолиз в практике врача-дерматовенеролога

Автор: Шустова О.А., Бобко Н.К.
Журнал: Саратовский научно-медицинский журнал @ssmj
Рубрика: Кожные болезни
Статья в выпуске: 3 т.10, 2014 года.
Бесплатный доступ
В статье описан клинический случай редкого дерматоза — врожденного дистрофического буллезного эпидермолиза у ребенка 9 лет.
Буллезный эпидермолиз, клиника
Короткий адрес: https://sciup.org/14918006
IDR: 14918006
Текст научной статьи Врожденный буллезный эпидермолиз в практике врача-дерматовенеролога
1 Введение. Буллезный эпидермолиз — это группа редких наследственных заболеваний, включающая около 30 форм, характеризующихся нарушением межклеточных контактов в эпидермисе или дерме, что при малейшей травме приводит к образованию пузырей [1, 2]
С учетом клинических особенностей и методов электронной микроскопии выделяют три основные группы врожденного буллезного эпидермолиза: простой буллезный эпидермолиз, пограничный буллезный эпидермолиз и дистрофический буллезный эпидермолиз [3].
При простой форме буллезного эпидермолиза причиной возникновения внутриэпидермальных пузырей служит мутация генов, кодирующих синтез кератина 5, 14 типов и плектина. Она приводит к дестабилизации сети тонофиламентов и цитолизу кератиноцитов базального слоя, в результате чего базальный слой отслаивается, при этом неповрежденная базальная мембрана находится в основании пузыря [4, 5].
Наследуются чаще всего по аутосомно-доминантному типу [4].
Простой буллезный эпидермолиз начинается с рождения или в первые дни жизни. Заболевание характеризуется появлением пузырей на местах механической травмы (локти, колени, кисти, стопы, поясница). Пузыри имеют различные размеры, прозрачное, редко — геморрагическое содержимое. Эрозии, образующиеся после вскрытия пузырей, быстро эпителизируются, не оставляя следов. Слизистые оболочки поражаются очень редко. В патологический процесс ногтевые пластинки обычно не вовлекаются [1, 2].
При пограничной форме буллезного эпидермолиза пузыри образуются в результате расщепления светлой пластинки базальной мембраны на границе эпидермиса и дермы. Генетический дефект — мутации генов, кодирующих антиген буллезного пемфигоида (в гемидесмосомах) и ламинин 332 (в якорных филаментах). Плотная пластинка базальной мем-
браны находится в основании пузыря. Тип наследования аутосомно-рецессивный [2, 4, 5].
К типичным симптомам данной формы относятся образование множества пузырей, эрозий и атрофических рубцов кожи, ониходистрофия, приводящая к полной утрате ногтевых пластин, тяжелое поражение мягких тканей в ротовой полости, гипоплазия эмали и тяжелый кариес. Патогномоничным симптомом является обильная грануляционная ткань, симметрично образующаяся вокруг рта, в области средней части лица и вокруг носа, в верхней части спины, подмышечных впадинах и ногтевых валиках. Возможными системными осложнениями являются тяжелая по-лиэтиологическая анемия, задержка роста, эрозии и стриктуры желудочно-кишечного тракта, поражение слизистых оболочек верхних дыхательных путей и мочеполового тракта, поражение почек, наружных оболочек глаза [6].
При дистрофическом буллезном эпидермолизе пузыри формируются глубоко под базальной мембраной, поэтому после заживления остаются рубцы. Развитие данной формы обусловлено мутацией гена, кодирующего коллаген VII типа — компонент крепящихся фибрилл. Из-за этого нарушения крепящиеся фибриллы рудиментарны или отсутствуют [2, 4, 5].
Описаны как аутосомно-рецессивные, так и аутосомно-доминантные варианты наследования дистрофического буллезного эпидермолиза [4].
Доминантный дистрофический буллезный эпидермолиз начинается с рождения или первых дней жизни. В первые месяцы поражение кожи генерализованное, в дальнейшем пузыри возникают обычно на одних и тех же часто травмируемых участках: кистях, стопах, коленях, локтях, шее. Заживление происходит с образованием атрофического рубца. Ногтевые пластинки поражены у всех больных, и лишь в редких случаях ногти отсутствуют, чаще они дистрофичны. Рост и развитие детей не нарушены. С возрастом пузыри появляются все реже, и у взрослых о наличии болезни могут напоминать только дистрофические изменения ногтей и едва заметные рубцы на локтях, коленях и лодыжках [3, 7].
Рецессивный дистрофический буллезный эпидермолиз протекает тяжело, часто приводит к смерти в раннем возрасте. Заболевание всегда возникает с рождения или первых часов жизни. Уже при рождении часто эрозирована кожа конечностей.
В первые дни жизни происходит распространение высыпаний. Заживление происходит с образованием атрофических рубцов, на кистях и стопах постепенно развиваются контрактуры и синдактилии. Ногтевые пластинки отсутствуют с рождения или постепенно утрачиваются в результате образования подногтевых пузырей. На слизистой оболочке полости рта, пищевода, прямой кишки также возникают множественные пузыри. Процесс рубцевания во рту приводит к ограничению подвижности языка, атрофии его сосочков, заращению вестибулярных складок и микростомии, в пищеводе — к его сужению, нарушению проходимости пищи, в прямой кишке — к хроническим запорам, резким болям при дефекации. Зубы поражены у всех больных, преобладают кариес, дефекты зубной эмали [1, 3, 6].
Этиотропного лечения буллезного эпидермолиза пока нет. Поэтому лечение больных является симптоматическим. Выбор симптоматических методов зависит от тяжести и обширности поражения. Образование пузырей может быть минимизировано ограничением травматических воздействий и использованием мягкой, хорошо подобранной обуви, одежды [2, 4, 7].
Описание клинического случая. Пациент А., 9 лет, состоит на диспансерном учете в ГУЗ « СОКВД» с 2005 г. с диагнозом: «Врожденный буллезный эпидермолиз».
Родился от пятой беременности, вторые срочные роды. В родах 2-кратное обвитие пуповины. Масса тела при рождении 3 кг 450 г, рост 52 см. С рождения состояние тяжелое за счет поражения кожи. Кожный процесс носил распространенный характер: на коже туловища, верхних, нижних конечностей имелись множественные пузыри с серозно-геморрагическим содержимым, склонные к периферическому росту и слиянию. На месте вскрывшихся пузырей формировались обширные эрозии, с сочным ярко-красным дном. С рождения укорочение правой нижней конечности, ониходистрофия. В тяжелом состоянии переведен в отделение реанимации новорожденных, где было выполнено гистологическое исследование кожи, на основании которого выставлен диагноз: «Пограничный буллезный эпидермолиз тип Херлит-ца». По мере роста ребенка пузыри появлялись в основном в местах, подверженных механическому воздействию (голени, колени, предплечья, локти), а также могли возникнуть на любом участке кожного покрова (рис. 1).
С двухлетнего возраста отмечалось некоторое улучшение кожного процесса, высыпания в основном локализуются на шее, паховых областях.
С 2006 г. состоит на диспансерном учете по месту жительства у гастроэнтеролога с диагнозом: «Сужение верхней трети пищевода, реактивные изменения поджелудочной железы, колодискинезия по гипомо-торному типу»; у ортопеда с диагнозом: «Гипоплазия костей правой стопы, варусная установка правой стопы, вальгусная установка левой стопы».
В 2013 г. в Чешской Республике, в Университетской больнице г. Брно, исследовали биоптат кожи ребенка (методом антигенного картирования), а также был произведен ДНК-молекулярный анализ.
Антигенное картирование: белки кератин 5, кератин 14, коллаген XVII, ламинин 332: окрашиваются обычно, коллаген VII типа: окрашивается слабо.
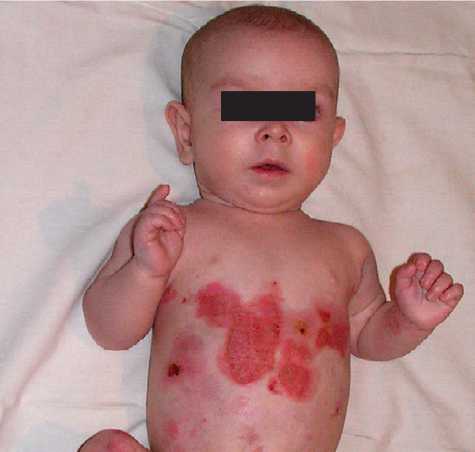
Рис. 1. Пациент А, 4 мес.
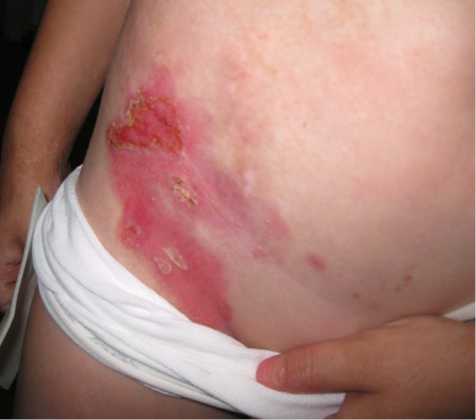
Рис.2. Пациент А., 9 лет
ДНК-молекулярный анализ: у пациента обнаружены 2 мутации в гене, отвечающие за синтез коллагена VII типа.
На основании анамнеза, клинической картины, лабораторных данных ребенку был выставлен диагноз: «Дистрофический буллезный эпидермолиз, аутосомно-рецессивный тип».
На сегодняшний день пузыри в основном локализуются в паховых областях, ладонной поверхности кистей. Кожный процесс представлен очагами эритемы ярко-розового цвета, с неровными, нечеткими контурами, на поверхности эрозии, с обрывками эпидермиса по периферии, местами покрытые серозногеморрагическими корочками. (рис. 2). На коже груди, живота, верхних, нижних конечностях множественные атрофические рубцы, гиперпигментация.
Ногтевые пластинки на правой руке отсутствуют на I и III пальцах, на левой руке на III пальце. На нижних конечностях ногтевые пластинки отсутствуют полностью.
За время диспансерного наблюдения пациент получает симптоматическую терапию, направленную на предупреждение инфекционных осложнений со стороны кожи и стимуляцию репаративных процессов.
Заключение. Приведенное клиническое наблюдение интересно из-за редкой встречаемости данного дерматоза, трудности диагностики и отсутствия эффективных методов лечения. К сожалению, буллезный эпидермолиз — неизлечимое заболевание, однако это совсем не означает, что таким пациентам нельзя помочь. Основным в лечении болезни является правильный уход за кожей, который позволяет минимизировать осложнения и адаптировать таких людей в обществе. Необходимо отметить, что диспансерное наблюдение пациентов с данным заболеванием должно осуществляться в течение всей жизни.
Список литературы Врожденный буллезный эпидермолиз в практике врача-дерматовенеролога
- Альбанова В.И. Наследственная пузырчатка (бул-лезный эпидермолиз). Русский медицинский журнал 1997; 5 (11): 735-744
- Вульф К., Джонсон P., Сюрмонд Д. Дерматология: Атлас-справочник. М.: Практика, 2007; с. 138-153
- Родионов В.Г., Провизион Л.Н. Дистрофический буллезный эпидермолиз (клиническое наблюдение). Перспективы медицины и биологии 2012; 4 (1): 91-94
- Кей Шу-Мей Кейн, Питер А.Лио. Детская дерматология: Цветной атлас и справочник. М.: Бином, 2011; с. 94-104
- Альбанова В.И., Гольченко В.А. Наследственный буллезный эпидермолиз: современные представления об этиологии и патогенезе. Российский журнал кожных и венерических болезней 2013; (2): 15-19
- Махнева H.B., Андреева Т.Е. Случай тяжелого генерализованного рецессивно-дистрофического врожденного буллезного эпидермолиза. Российский журнал кожных и венерических болезней 2012; (3): 12-17
- Калюжная Л.Д. Тактика ведения больных с буллезным эпидермоли-зом. Украинский журнал дерматологии, венерологии, косметологии 2003; (4): 27-29.
