Вычисление сеток взаимодействия молекул с использованием графических процессоров

Автор: Фарков Михаил Александрович
Журнал: Космические аппараты и технологии.
Рубрика: Информационные технологии
Статья в выпуске: 3-4 (5-6), 2013 года.
Бесплатный доступ
Описана теоретическая база молекулярного лиганд-белкового докинга; описан сеточный подход к ускорению докинга; представлена декомпозиция задачи для параллельных вычислительных систем; обоснована актуальность применения графических процессоров.
Молекулярный докинг, виртуальный скрининг, лиганд-белковый докинг
Короткий адрес: https://sciup.org/14117308
IDR: 14117308 | УДК: 004.42
The calculation of grids of molecules interaction using graphics processors
Theoretical base of molecular ligands-proteins docking was described. Grids approach for molecular docking was described. Task's decomposition for parallel computation systems was presented. The relevance of using GPU for solving problem was proved.
Текст научной статьи Вычисление сеток взаимодействия молекул с использованием графических процессоров
M. A. Farkov
Siberian Federal University , Krasnoyarsk , Russia
THE CALCULATION OF GRIDS OF MOLECULES INTERACTION USING GRAPHICS PROCESSORS
Theoretical base of molecular ligands-proteins docking was described. Grids approach for molecular docking was described. Task's decomposition for parallel computation systems was presented. The relevance of using GPU for solving problem was proved.
GPGPU , CUDA , molecular docking , virtual screening , ligand-protein docking.
Одной из ключевых проблем при разработке новых лекарств является подбор перспективных химических соединений (называемых лигандами), кандидатов в лекарства. Этот процесс может занимать существенное время в процессе разработки (от года до трёх лет) и, кроме того, является достаточно затратным (траты на реагенты и высокоточное оборудования для проведения реакций синтеза) [1]. Помимо этого ошибки, допущенные на данном этапе, то есть отбор соединений, не оказывающих необходимого биологического отклика или приводящих к значительным побочным эффектам, чреваты катастрофическими финансовыми и временными потерями на последующих этапах (доклинических и клинических испытаниях). Для ускорения, а также снижения финансовых и временных затрат применяют компьютерное моделирование, называемое молекулярным докингом.
Молекулярный лиганд-беловый докинг – это моделирование процесса взаимодействия биомишени (как правило, белка) с лигандом (небольшим молекулярным соединением). Выполнение молекулярного докинга позволяет определить принципиальную возможность протекания химической реакции между молекулами, а также оценить энергию реакции с целью выделить наиболее перспективные лиганды для последующих испытаний in vitro, а затем и in vivo.
Выполнение докинга - достаточно вычислительно затратная процедура и, как правило, является компромиссом между точностью и скоростью вычислений. Для ускорения процедуры докинга достаточно часто используются многоядерные, а также распределённые вычислительные системы, вместе с тем гетерогенные вычислительные системы, в состав которых входят графические процессоры, применяются для лиганд-белкового докинга необоснованно редко.
Энергия взаимодействия молекул может быть рассчитана как линейная комбинация нескольких компонент, которые условно можно разделить на две подгруппы межмолекулярного и внутримолекулярного взаимодействия [2; 3; 4]:
Etotal Evdw + Eelec + Ebond + Eangle + Etorsion, где к межмолекулярному взаимодействию относятся Evdw, Eelec, энергия ван-дер-вааль-сового взаимодействия (между атомами 2, 5 на рис. 1) и энергия электростатического взаимодействия (между атомами 1,6 на рис. 1) соответственно. К внутримолекулярному взаимодействию относятся: энергия взаимодействия между двумя ковалентно-связанными атомами Ebond (атомы 2, 3 на рис. 1); энергия взаимодействия между тремя ковалентносвязанными атомами Eangle (атомы 2, 3, 4 на рис. 1); энергия взаимодействия между атомами, разделёнными тремя ковалентными связями и образующими торсионный угол Etorsion (атомы 1, 2, 3, 4 на рис. 1).
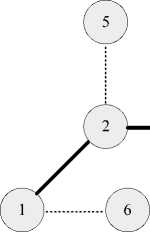
+
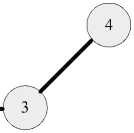
Ковалентная связь
Межмолекулярное взаимодействие
Рис. 1. Атомы в молекулах
Наиболее существенный вклад в общую энергию оказывают энергии ван-дер-вааль-сового и электростатического взаимодействия. При этом, в свою очередь, каждая из этих компонент является линейной комбинацией взаимодействия каждого атома лиганда с каждым атомом биомишени. Энергия ван-дер-вааль-сового взаимодействия оценивается согласно потенциалу Леннарда-Джонсона:
^Sij ((Г0ij IГу)12 - 2(r0ij IГу)6), где εij и r0ij – константы; rij – расстояние между взаимодействующими атомами [5; 6]. Энергия электростатического взаимодействия оценивается согласно закону Кулона:
Σ(qi qj) / (εrij ), где s - диэлектрическая проницаемость среды; Гу - расстояние между взаимодействующими атомами; qi, qy - заряды атомов [7; 8]. Тот факт, что каждая из этих компонент является суммой взаимодействия отдельных атомов молекул, лежит в основе сеточного подхода к ускорению выполнения молекулярного докинга. Кроме того, при переборе ориентаций лиганда относительно интересующей области (называемой сайтом связывания) молекулы биомишени, одни и те же типы атомов лиганда будут достаточно часто попадать при- 47
близительно в одни и те же точки пространства. На этих двух идеях основывается сеточный подход к ускорению докинга, согласно которому некоторая часть биомишени помещается в ограниченную область, например прямоугольный параллелепипед, и полагается неподвижной. После чего для каждой точки пространства в пределах сетки с некоторым шагом вычисляются компоненты энергии для всех типов атомов (называемых пробами). представленных в лиганде. В результате на выходе данной процедуры получается набор сеток, которые в дальнейшем используются для определения оптимальной конформации биомишени и лиганда.
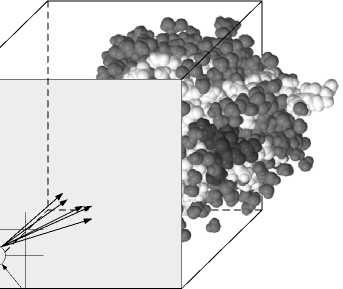
Атом проба
Рис. 2. Пример сетки
Преимущество такого подхода заключается в том, что при проверке большого количества расположений лиганда в сайте связывания биомишени нет необходимости повторять значительное количество однотипных вычислений (объём которых линейно увеличивается с ростом количества атомов биомишени). Требуемые значения берутся из пред-вычисленных сеток. При выполнении гибкого лиганд-белкового докинга с учётом подвижности лиганда данный подход даёт ещё большее преимущество, так как возрастает коли-
ИССЛЕДОВАНИЯ
HAVKO____________ Ж ГРАДА
чество степеней свободы лиганда, по которым осуществляется его позиционирование (к 6 стандартным степеням свободы добавляются внутренние степени свободы лиганда: вращение вокруг ковалентных связей и изменение валентных и торсионных углов).
Следует отметить, что вычисление сеток возможно только для энергий ван-дер-вааль-сового и электростатического взаимодействия, так как при их расчёте нет необходимости задавать определённое положение лиганда в сайте 48 связывания биомишени. На входе процедуры расчёта сеток имеется множество биомишеней TARGETS, где каждому элементу targets i соответствует множество лигандов LIGANDS i , где каждому лиганду ligand - соответствует множество типов атомов ATOM_TYPES ij , такое, что каждый тип atom_type - k хотя бы один раз присутствует в лиганде. В результате вычислений необходимо получить некоторое множество сеток GRIDS, элемент которого grid ijk является результатом вычисления сетки силового поля для атома типа k , принадлежащего лиганду j и взаимодействующего с белком i . Так как справедливо, что сетки силового поля для одного типа атомов, присутствующего в различных лигандах и взаимодействую щ их с одной и т ой же биомишенью, являются эквивалентными, то есть
№ 3-4 (6) июль-декабрь 2013
типов атомов [9; 10; 11]. Кроме того, при вычислении достаточно больших сеток данный подход не оправдывает себя и с точки зрения скорости вычислений.
Отдельно стоит рассмотреть вычисление сеток для электростатического взаимодействия. Нетрудно заметить, что с целью дополнительного упрощения можно вынести заряд атома лиганда из процесса вычислений. В результате необходимо будет вычислить только одну сетку электростатического взаимодействия для биомишени. По аналогии можно вынести и диэлектрическую проницаемость среды, но в работе используется дистанционно зависимый подход к расчёту диэлектрической проницаемости среды, который даёт более точные результаты, сравнимые с результатами, получаемыми при выполнении расчётов методами молекулярной динамики, при значительно меньших вычислительных затратах [12].
В результате для каждой биомишени targetsi необходимо вычислить
| ATOM_TYPESi0 u ATOM_TYPES i1 u ...
S„ и ... и ATOM_TYPES ,(N-,)| + 1,
N: = |LIGANDSi| atomjypes jk =
= atomjypesimn ^ gridyk = gridimn, то можно уменьшить количество вычислений, заменив попарное вычисление сеток для каждой биомишени targetsi из множества TARGETS с соответствующими ей лигандами из множества LIGANDSi, на вычисления сеток для биомишени i, и типов атомов, присутствующих хотя бы в одном лиганде из множества LIGANDSi. Следует отметить, что в некоторых случаях для вычисления сеток нет необходимости получать информацию о типах атомов лиганда, а целесообразнее (с точки зрения скорости работы) вычислять сетки для всех типов атомов, присутствующих в силовом поле. Несмотря на это, поскольку одной из целей разрабатываемого программного обеспечения является максимальная взаимозаменяемость используемого силового поля и по умолчанию используется точное силовое поле GAFF (general amber force field), такой подход не рационален ввиду наличия достаточно большого количества обрабатываемых сеток.
Поскольку энергия взаимодействия в некоторой точке пространства не зависит от значения энергии в окружающих её точках пространства как для ван-дер-ваальсового взаимодействия, так и для электростатического взаимодействия, процесс вычислений отдельной сетки имеет высокую степень параллелизма по данным. Фактически значение энергии взаимодействия для каждой точки сетки вычисляется независимо. Кроме того, дополнительно следует отметить, что вычисления отдельных сеток, даже для одинаковой биомишени, также полностью независимы. Наилучшим образом для подобных задач, обладающих высоким параллелизмом по данным, подходят графические процессоры, поскольку позволяют выполнять значительное количество однотипных вычислений над данными физически параллельно в рамках SIMD (single instruction multiple data) модели вычислений. Принимая во внимание сказанное, можно выполнить двухуровневую декомпозицию задачи для выполнения параллельных вычислений: первый уровень – уровень отдельных сеток; второй – уровень отдельных точек сетки. Применяя такой вариант декомпозиции для графических процессоров, на первом уровне можно балансировать вычислительную нагрузку между несколькими графическими процессорами в составе одного компьютера. Разбиение же на вычисления отдельных точек сетки можно производить для одного графического процессора, так как графический процессор имеет значительное количество простых SIMD ядер, что позволяет вычислять большое количество точек сетки физически параллельно. Вычислительная сложность процедуры расчёта одной сетки для одной биомишени равна O(n·m), где n количество ячеек в сетке, а m – количество атомов в биомишени. Количество ячеек может достаточно произвольно меняться в зависимости от исследуемой области биомишени и требуемой точности, но, как правило, находится в диапазоне от 64 000 (сетка размерностью 40×40×40) до 1 000 000 (сетка размерностью 100×100×100). Так как вычислительная нагрузка велика, алгоритм является достаточно масштабируемым, вследствие чего рост количества отдельных вычислителей не приведёт к их простою, а из-за специфики диспетчеризации отдельных нитей на графических процессорах увеличение параллельно работающих вычислителей не приведёт к существенному росту накладных расходов на запуск вычислений. Кроме того, повышению масштабируемости алгоритма способствует отсутствие необходимости выполнять какую-либо синхронизацию вычислений отдельной сетки в процессе работы.
Список литературы Вычисление сеток взаимодействия молекул с использованием графических процессоров
- Kuntz I.D. Structure-based strategies for drug design and discovery // Science. 1992 by the American Association for the Advancement of Science, 1992. Vol. 257, №5073. P. 1078-1082.
- Natasja Brooijmans, Irwin D. Kuntz. Molecular recognition and docking algorithms. Annu. Rev. Biophys. Biomol. Struct. 2003. 32:335-73.
- MacKerell A.D., Jr. «Atomistic Models and Force Fields» in Computational Biochemistry and Biophysics, O.M. Becker, A.D. MacKerell, Jr., B. Roux and M.Watanabe, Eds., Marcel Dekker, Inc. New York, 2001, p. 7-38.
- MacKerell A.D., Bashford D., Dunbrack R.L., Evanseck J.D., Field M.J., Fischer S., Gao J. et al. (1998). All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. The Journal of Physical Chemistry B, 102(18), 3586-3616. DOI: 10.1021/jp973084f
- Goodford P.J. A computational procedure for determining energetically favorable binding sites on biologically important macromolecules. J Med Chem 1985; 28:849-857.
- Michael K. Gilson, Huan-Xiang Zhou. Calculation of Protein-Ligand Binding Affinities. Rev. Biophys. Biomol. Struct 2007. 36:21-42.
- Todd J.A. Ewinga, Shingo Makinoa, A. Geoffrey Skillmana, Irwin D. Kuntza. DOCK 4.0: Search strategies for automated molecular docking of flexible molecule databases. Journal of Computer-Aided Molecular Design, 15: 411-428, 2001.
- Cornell W.D., Cieplak P., Bayly C.I., Gould I.R., Merz K.M., Ferguson D.M., Spellmeyer D.C. et al. (1995). A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. (P.E. Bourne, Ed.) Journal of the American Chemical Society, 117(19), 5179-5197.
- Wang J., Wang W., Kollman P.A. & Case D.A. (2006). Automatic atom type and bond type perception in molecular mechanical calculations. Journal of molecular graphics modelling, 25(2), 247-260.
- Wang J., Wolf R.M., Caldwell J.W., Kollman P.A., Case D.A. Development and testing of a general AMBER force field. Journal of Computational Chemistry, 25, 2004, 1157-1174.
- Wang J. et al. Antechamber, An Accessory Software Package For Molecular Mechanical Calculations // Molecules. AMER CHEMICAL SOC, 2001. Vol. 222, №2. P. U403-U403.
- Mehler E.L. & Solmajer T. (1991). Electrostatic effects in proteins: comparison of dielectric and charge models. Protein Engineering, 4(8), 903-910.
