Взаимодействие цинк-тетрафенилпорфина, бромистого пропаргила и пероксидазы хрена с анилинами

Автор: Андреев Владимир Петрович, Соболев Павел Сергеевич, Зайцев Дмитрий Олегович, Галибина Наталья Алексеевна, Зыкина Наталья Сергеевна, Плясунова Любовь Юрьевна, Романова Марина Игоревна
Журнал: Ученые записки Петрозаводского государственного университета @uchzap-petrsu
Рубрика: Биология
Статья в выпуске: 6 (119), 2011 года.
Бесплатный доступ
Порфирины, координация, нуклеофильное замещение, пероксидаза хрена, гетероароматические n-оксиды
Короткий адрес: https://sciup.org/14749971
IDR: 14749971
Текст статьи Взаимодействие цинк-тетрафенилпорфина, бромистого пропаргила и пероксидазы хрена с анилинами
В конце XIX - начале XX века работы одного из основателей российской биохимии академика А. Н. Баха (в частности, формулировка и экспериментальное доказательство совместно с Р Шодой перекисной теории биологического окисления) получили всемирное признание. В 1903 году ими были выделены частично очищенные препараты пероксидазы из корней хрена, содержащие две активные фракции. Первая фракция получила название оксигеназы, а вторая - пероксидазы [8]. Гомогенный же препарат был получен несколько десятилетий спустя Вильшаттером и Теореллом [9].
Различные схемы протекания пероксидазной реакции предполагают взаимодействие прото© Андреев В. П., Соболев П. С., Зайцев Д. О., Галибина Н. А., нов субстрата с дистальным остатком Arg, присутствующим во всех пероксидазах, последовательности которых определены к настоящему времени. Однако проблема субстратной специфичности классических пероксидаз растений до сих пор не решена. Каждый фермент имеет свой собственный профиль субстратной специфичности, и пока, к сожалению, нельзя заранее предсказать, какова будет его активность по отношению к выбранному донору электронов [9].
В настоящее время для описания взаимодействия пероксидазы хрена (HRPC) с субстратами, подобными фенолам и анилинам, широко используется механизм, предложенный Родригес-Лопес [28]:
Зыкина Н. С., Плясунова Л. Ю., Романова М. И., 2011
Ввиду того что способность к окислению анилинов и фенолов должна быть напрямую связана с электронодонорными и акцепторными свойствами заместителей в ароматическом кольце, ряд авторов для количественного описания реакционной способности пероксидазы хрена и ее окисленных форм по отношению к этим субстратам использовали уравнение Гаммета и его модификации [10]:
lg (k x/kH ) = ро, где kx и kн обозначают константы скоростей реакции замещенного и незамещенного соединений соответственно, о - определяемая природой и положением заместителя в бензольном кольце константа, характеризующая его способность изменять электронную плотность, р - параметр, отражающий чувствительность процесса к смене заместителя.
В случае окисления фенолов и анилинов посредством cpd-I и cpd-II обнаружены, с одной стороны, линейные зависимости между логарифмами констант скорости этой реакции, с другой - с о-значениями Гаммета заместителей в бензольном кольце [22], [25], а также потенциалами ионизации высшей занятой молекулярной орбитали [16], [29] некоторых фенолов и анилинов, причем скорость окисления фенолов посредством cpd-II является на 1-3 порядка более высокой, чем для анилинов с близкими потенциалами ионизации [16], [29], [30]. Л. Фенолл и соавторы [23] проанализировали механизм взаимодействия этих субстратов с сpd-II, включающий перенос электрона (окисление) субстрата и его дальнейшее депротонирование. Авторы делают вывод, что скорость реакции главным образом контролируется потенциалом полуволны электрохимического окисления (Е1/2) субстрата в пределах одного класса соединений, но субстраты различных классов с близкими Е1/2 могут окисляться с очень сильно различающимися скоростями (как, например, 4-метилфенол и 4-метил-анилин, имеющие одно и то же значение Е1/2 -0,87 вольт). Тот же самый эффект наблюдается при окислении серии фенолов посредством cpd-I или cpd-II [21], [24]. Следует отметить, что константы скорости реакции анилинов с соединением II пероксидазы примерно на порядок ниже, чем для реакции с соединением I.
Нами обнаружено, что, как и в случае гетеро -ароматических N-оксидов, а также 3- и 4-заме-щенных пиридинов [1], [3], [4] (при отсутствии стерических факторов), в электронных спектрах поглощения (ЭСП) смещения максимумов полос (АХ) Zn-ТФП в хлороформе при взаимодействии с анилинами (Iа-г) линейно коррелируют с логарифмами констант устойчивости (К) комплексов, c pKa лигандов в воде и о-константами заместителей в бензольном кольце (табл. 1).
Мы полагаем, что полученные нами константы устойчивости анилинов с Zn-ТФП относятся к молекулярным комплексам n, v-типа состава 1:1 с донорно-акцепторной связью N - Zn.
х
nh2 la - г
X = Н (а); 4-Ме (б); 4-ОМе (в); З-Ме (г);
В частности, линейная корреляция между pKa в воде и lgK в хлороформе (табл. 1) подтверждает, что протонирование и координация лигандов (Iа-г) с металлопорфирином осуществляется c участием одного и того же центра - атома азота аминогруппы. На рис. 1 представлены данные рентгеноструктурного анализа (РСА) относительно структуры продуктов взаимодействия 2-хлор- и 3-нитроанилина с Zn-ТФП состава 1:1.
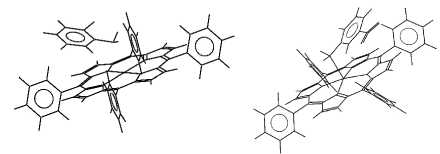
а б
Рис. 1. Строение комплексов Zn-ТФП состава 1:1 с 3-нитроанилином (HAMLAI) (а) и с 2-С1-анилином (JIVNIL) (б) согласно данным РСА (аббревиатура в скобках соответствует ссылке (CSD refcode), используемой в Cambridge Structural Database [20])
Таблица 1
Константы устойчивости (К) молекулярных комплексов Zn-ТФП с анилинами в хлороформе при 25 °C, смещение максимума полосы поглощения II (АХ II ) в спектре Zn-ТФП при комплексообразовании, константы скорости (k) реакции окисления анилинов соединением II пероксидазы хрена (схема, уравнение (3)) в фосфатном буфере, pH 7,0 [29], о - и о + - константы Гаммета и Брауна и pKa анилинов в воде при 25 ° С [16]
lgK = -0,45g+ + 2,18 r = 0,997 lgk = 2,31g+ + 5,04 r = 0,994
AXII = 3,66 lgK + 6,21 r = 0,966 lgk = - 1,28 AXII - 13,05 r = 0,965 lgk = -4,99 lgK - 5,80 r = 0,994
Нами обнаружено, что, в отличие от пиридинов и гетероароматических N-оксидов [1], [4], поведение анилинов при координации с Zn-ТФП значительно лучше описывается с использованием G+-констант Брауна заместителей (табл. 1, r = 0,997), а не G-констант Гаммета (r = 0,960). В монографии А. С. Днепровского указывается, «что необходимость применения G+-констант обусловлена наличием именно свободной орбитали на реакционном центре, а не положительного заряда» [10]. Подчеркнем, что в Zn-ТФП остаточный положительный заряд на атоме цинка слишком мал для того, чтобы сильно поляризовать молекулы анилинов за счет кулоновского взаимодействия.
Между AX II и lgK также наблюдается хорошая линейная зависимость (табл. 1, r = 0,966).
Ввиду подобия строения комплексов Zn-ТФП с лигандами и переходных состояний в реакциях S N [5] с последними в роли нуклеофилов мы попытались найти количественные соотношения между физическими параметрами, характеризующими эти процессы.
Оказалось, что и константы скорости реакций нуклеофильного замещения k с анилинами в смесях метанол-ацетонитрил (50-100 %) при 35 °С [27]
-
— жда+вдед + ^S^Z
X = н, Me, 4-OMe, 4-Cl, 3-NO 2 ;
Y = H, 4-Cl; Z = H, 4-Me, 4-Cl, 3-NO 2 , и константы k 2 , kXY, kXZ аминолиза бензойных ангидридов в метаноле при 25-45 °С [26],
^XY
-^— XC6H4NHCOCeH4Y + XC6H4NH3+ + CeH5COO-
2XCeH4NH2 + Y(X)C6H4COOCOCeH5
XCeH4NHC0CeHg +xc6h4nh3+ +zc6h4coo-
X = H, 4-OMe, 4-Me, 4-Br, 3-Cl;
-
Y(Z) = H, 4-OMe, 4-Me, 3-Cl, 4-NO 2 , и константы скорости реакции транс-З-нитро-фенил-в-хлорвинилкетона с анилинами (X = = 4-NH 2 , 4-MeO, 4-Me, 4-Br, 3-Cl, 3-NO 2 ) в изопропиловом спирте при 25 °С [12] также линейно коррелируют (r = 0,97-0,99) с константами комплексообразования анилинов с Zn-ТФП в хлороформе при 25 °С и смещениями максимумов полос поглощения (AX) МП.
Таким образом, между кинетическими параметрами комплексообразования Zn-ТФП и некоторых реакций нуклеофильного замещения (константы устойчивости комплексов (К), величины смещения максимумов полос поглощения МП (AX), основность лиганда / нуклеофила / субстрата (pKa), константы скоростей реакций, g- и G+-константы заместителей в бензольном кольце) с участием анилинов существуют линейные зависимости.
Мы решили проверить, будут ли выполняться подобные корреляции и с ферментативными процессами на примере взаимодействия анилинов с пероксидазой хрена, содержащей гем в качестве простетической группы. Оказалось, что для стадии взаимодействия соединения II пероксидазы хрена (k в табл. 1; схема, уравнение (З); [29]) с анилинами, содержащими заместители в положении 3 и 4 бензольного кольца, наблюдаются линейные зависимости между lg K, lg k, pK a , g+ и AX (рис. 2 а, б), причем скорость этой ферментативной реакции, как и координация Zn-ТФП, лучше описывается с использованием G+-констант.
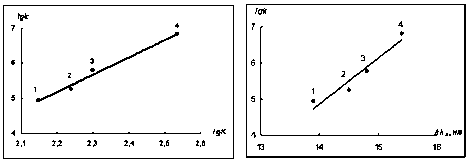
а б
Рис. 2. Зависимость lg k реакции окисления анилинов соединением II пероксидазы хрена от lg K (а, r = 0,994) и AX II (б, r = 0,965) процесса координации Zn-ТФП с анилинами; заместители: (1) 4-Н; (2) З-Me; (3) 4-CH 3 ; (4) 4-OCH 3
Следовательно, на основании численных значений К и AX (в соответствии с введенной нами шкалой нуклеофильности / основности [2]) для процесса координации Zn-ТФП с различными типами лигандов (координационная химия) можно предсказывать скорость как реакций нуклеофильного замещения (органическая химия), так и ферментативных реакций (биологическая химия) в различных органических и водных растворах, и наоборот.
В дальнейшем мы предполагаем детально разобраться в границах применимости предложенной нами новой шкалы.
Особое положение среди анилинов занимают диметиламиностирильные производные гетеро-ароматических N-оксидов, которые в органических растворителях образуют молекулярные комплексы с v-акцепторами (Zn-ТФП, BF 3 , ZnCl 2 , Н+) состава 1:1 с участием кислорода группы N—O, но в воде первое протонирование этих соединений осуществляется по аминогруппе. Комплексы же состава 1:2 (взаимодействие с N— O и N(CH3)2 группами), образующиеся при избытке кислоты Льюиса, неустойчивы и легко теряют вторую молекулу акцептора [1], [2], [4], [5].
Ранее в ПетрГУ А. Г. Анисимовым было обнаружено, что пероксидаза хрена окисляет N-оксид 4-(4-диметиламиностирил) пиридина (4-DPyO, обладает апоптогенной активностью), но механизм и продукты реакции им не были изучены.
Мы решили исследовать данный ферментативный процесс, для чего были синтезированы следующие стирильные производные N-оксидов пиридина и хинолина:
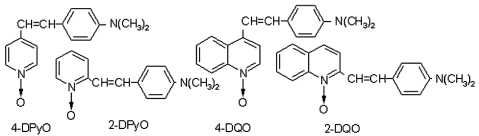
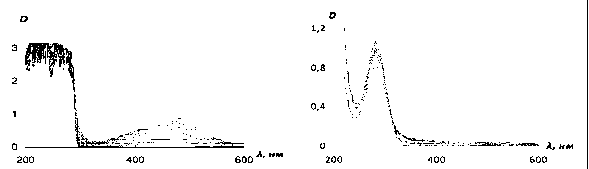
Скорость и направление их превращения под действием пероксидазы хрена контролировали методом электронной спектроскопии. На рис. 3 приведены электронные спектры поглощения (ЭСП) пероксидазы хрена и стирильных производных гетероароматических N-оксидов (4-DPyO, 2-DFyO, 2-DQO и 4-DQO).
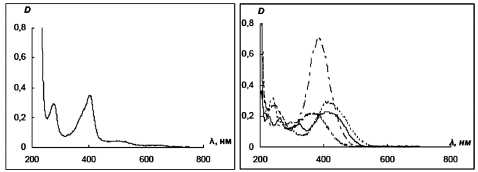
а б
Рис. 3. Электронные спектры в фосфатном буферном растворе pH 7,3-7,4: а) пероксидазы хрена (С = 0,7 мг/мл; RZ = D400/ D280 = 1,4); б) 4-DPyO (---), 2-DFyO (---), 2-DQO (-) и 4-DQO ( ); (С = 1 x 10 -5 моль/л, для 4-DPyO С = 2 х 10 -5 моль/л)
В ЭСП во время взаимодействия 4-DPyO с пероксидазой хрена в присутствии перекиси водорода в фосфатном буфере (рис. 4а) оптическая плотность уменьшается при 384 нм и увеличивается при 263 нм, и в течение 1 ч. субстрат практически полностью превращается в продукт(ы) реакции. Однако затем интенсивность поглощения света при 263 нм начинает медленно уменьшаться (10 % за первую неделю), указывая на дальнейшую трансформацию субстрата.
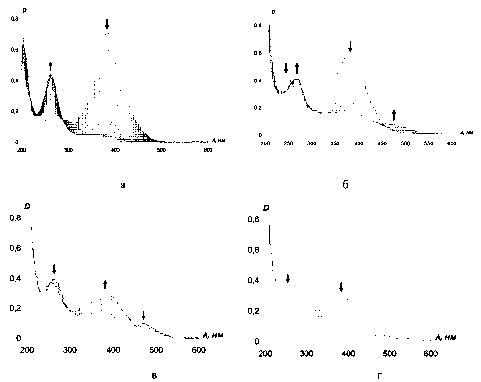
Рис. 4. Изменение ЭСП реакционной смеси, содержащей пероксидазу хрена, перекись водорода и 4-DPyO (в течение часа, (а)) или 2-DPyO (5-30 мин. (б), 30-60 мин. (в, г))
В отсутствие фермента происходят очень похожие, но значительно более медленные измене -ния ЭСП (за сутки интенсивность поглощения субстрата уменьшается на 70 %), то есть в данном случае пероксидаза лишь ускоряет процессы. Интересно, что фермент с 4-DPyO взаимо-действует и без добавления перекиси водорода, но реакция протекает еще медленнее, вероятно, за счет проявления пероксидазой хрена оксидазной активности и /или присутствия H 2 O 2 в растворе в следовых количествах. Последнее утверждение не лишено смысла, поскольку субстрат содержится в реакционной среде в очень низкой концентрации (10-5 М).
В случае 2-DPyO (имеет максимумы полос поглощения при 245 и 380 нм) наблюдается несколько иная и более сложная картина. В течение первых 5 мин. происходит увеличение оптической плотности при 380 нм и уменьшение при 245 нм, затем в течение получаса интенсивность этих полос поглощения снижается, но увеличивается оптическая плотность при 263 и 473 нм (рис. 4б). В течение следующих 0,5 ч. интенсивность поглощения света при 263 и 473 нм сначала падает с повторным ее увеличением при 380 нм (рис. 4в), но затем оптическая плотность всех полос поглощения уменьшается (рис. 4г) и ЭСП сглаживается. Мы предполагаем, что в данном случае наряду с химической реакцией в ЭСП регистрируется образование донорно -акцепторных комплексов фермента с различным количеством молекул субстрата, обладающих суммарно большей интенсивностью (но близкими максимумами) полос поглощения, чем исходный стирильный N-оксид.
Подчеркнем, что при взаимодействии 4-DPyO (в течение 1 ч.) и 2-DPyO (в течение 0,5 ч.) с пероксидазой хрена в ЭСП реакционной смеси наблюдаются изобестические точки при 225 и 290 нм для первого и при 250 и 300 нм для второго субстрата. Обычно при комплексообразовании их наличие интерпретируют как равновесие между двумя состояниями (свободным и связанным).
В нашем случае наличие изобестических точек можно отнести к существованию равновесия между стирильным N-оксидом и по крайней мере двумя продуктами реакции с фермент-суб-стратным комплексом в процессе, который сначала быстро и количественно приводит к образованию соединения А (кинетический контроль), а затем медленно - к соединению (или эквимо-лярной смеси) Б (термодинамический контроль).
При использовании в качестве субстрата 4-DQO изменения в ЭСП похожи на те, что происходят с 4-DPyO (изобестические точки при 270, 310 и 350 нм): в течение 1 ч. исчезают полосы поглощения субстрата (при 236 и 406 нм) и наблюдается появление максимума поглощения около 344 нм (рис. 5а), при этом происходит сме- щение крайнего слева максимума поглощения в коротковолновую область.
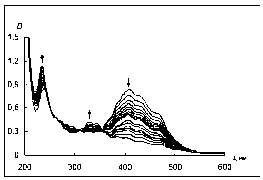
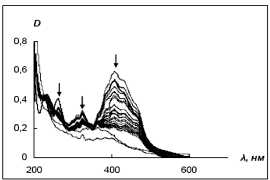
а б
Рис. 5. Изменение ЭСП реакционной смеси, содержащей пероксидазу хрена, перекись водорода и а) 4-DQO или б) 2-DQO в течение 4 суток
Наконец, в случае 2-DQO реакция заканчивается за 1-2 дня (рис. 5б), но протекает несколько иначе, чем с 4-DPyO, 2-DPyO и 4-DQO. В ЭСП реакционной смеси (в течение первых 3 мин. происходит увеличение оптической плотности при 408 и 325 нм, но резкое ее уменьшение при 262 нм) при этом исчезают 4 из 5 полос поглощения, характерных для субстрата, но увеличивается интенсивность полосы при 204 нм. Изобестические точки в ЭСП при взаимодействии данного субстрата с пероксидазой хрена отсутствуют, по -видимому, вследствие того, что продукты типа А и Б образуются с сопоставимыми скоростями и присутствуют в растворе одновременно.
С незамещенными стирильными аналогами (N-оксиды 4-стирилпиридина, 2- и 4-стирилхи-нолина), а также содержащими метокси- или нитрогруппы (N-оксиды 4-(4-метоксисти-рил)пиридина, 2-(2,4-диметоксистирил)- и 4-(4-нитростирил)хинолина), согласно данным ЭСП, пероксидаза хрена реагирует очень медленно, либо реакция практически не идет. Отметим, что во всех указанных случаях перекись водорода добавляли только один раз для инициации реакции.
Обычно пероксидазную активность в растительных тканях определяют с помощью метода электронной спектроскопии (в области 400800 нм) по накоплению окрашенных продуктов окисления орто-дианизидина, орто-фенилендиамина, 3,3′,5,5-тетраметилбензидина и других подобных соединений (используемые концентрации - порядка 10-4 моль/л [11]). Мы изучили взаимодействие пероксидаз, выделенных из ксилемы карельской узорчатой березы, на экзогенное соединение 4-DPyO. Оказалось, что, согласно данным ЭСП, направление этой ферментативной реакции носит такой же характер, что и с пероксидазой хрена (рис. 4а), но в условиях эксперимента (50 мМ Hepes, pH 7,5) реакция идет медленнее: через 90 мин. оптическая плотность субстрата при 384 нм уменьшается на 30 % (от 1,3 до 0,9), а продукта реакции при 263 нм достигает 0,5 единицы.
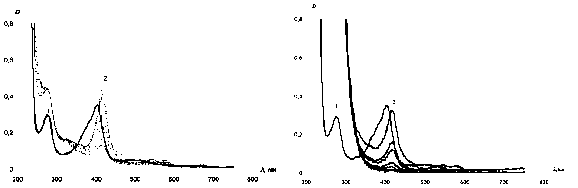
Для сравнения, ферментативная активность пероксидазы в тканях ксилемы была изучена на примере окисления классических субстратов гваякола (орто-метоксифенола) и бензидина (рис. 6 а, б). Как видно из представленных ЭСП, при окислении бензидина и гваякола требуются очень высокие концентрации субстрата для регистрации продуктов реакции области 400-800. Предложенная нами реакция окисления 4-DPyO является очень удобной для оценки ферментативной активности пероксидаз, поскольку позволяет одновременно оценивать концентрацию как субстрата, так и продукта реакции в экспериментах in vitro (рис 4а).
а б
Рис. 6. Изменение ЭСП реакционной смеси, содержащей ферментативный препарат из тканей ксилемы карельской березы, перекись водорода и а) гваякол (21,5 mM) б) бензидин (33,6 мкМ)
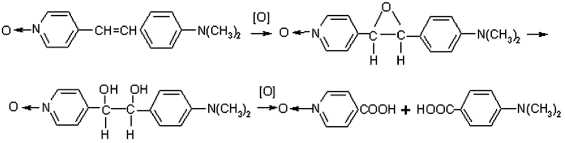
Ввиду того что пероксидазы способны катализировать реакции оксидазного, пероксидазного и оксигеназного окисления субстратов [7], [9], [13], [14], не исключено, что как взаимодействие с исходным субстратом, так и дальнейшая трансформация продуктов превращения стирильного N-оксида обусловлены реализацией всех трех типов активности фермента, причем по мере расходования перекиси водорода роль оксидазной и оксигеназной активности пероксидазы возрастает.
A priori можно предположить два основных направления окисления 4-DPyO (как и других стирильных N-оксидов).
-
1. По двойной связи стирильного фрагмента с образованием оксирана, гликоля и/или N-оксида изоникотиновой и 4-диметиламинобензойной (4-ДАБК) кислот, причем ДАБК может быть далее окислена до N-оксида 4-диметиламинобен-зойной кислоты
-
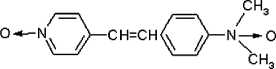
2. По атому азота диметиламиногруппы а) с образованием стирильного ди-N-оксида
-
б) или дальнейшим его окислением по двойной связи, как описано выше. Не исключено, что все эти процессы осуществляются параллельно.
Указанное направление реакции не противоречит ЭСП и временам удерживания (ВЭЖХ) предполагаемых продуктов превращений 4-DPyO
(N-оксида изоникотиновой кислоты и 4-ДАБК), использованных в качестве стандартов. Косвенным его подтверждением служит и то, что неферментативное окисление 4-DPyO перекисью водорода в кислой и щелочной среде дает аналогичные результаты.
С целью разработки метода, позволяющего облегчить количественное выделение продуктов реакции пероксидазы со стирильными производными N-оксидов пиридинов и хинолинов, мы исследовали взаимодействие фермента с 4-DPyO в водно-спиртовых безбуферных растворах (стирильные производные в спирте растворимы гораздо лучше, чем в воде). Оказалось, что в воде и 25 % (по объему) этаноле в результате реакции субстрат исчезает, как и в фосфатном буфере, в течение 1 ч., в 50 % этаноле - в течение суток, а в 75 и 96 % этаноле - за несколько суток расходуется наполовину; при этом, согласно ЭСП (возрастает интенсивность поглощения при 384 нм), с увеличением доли этанола время образования донорно-акцепторных комплексов фермента с молекулами субстрата возрастает от нескольких минут до нескольких часов.
Зная, что высокие концентрации H 2 O 2 могут приводить к быстрому разрушению пероксидазы хрена, мы проверили влияние различных концентраций перекиси водорода на устойчивость растворов фермента во времени.
а б
Рис. 7. Изменение ЭСП пероксидазы хрена во времени в зависимости от концентрации перекиси водорода (а - 0,002 %, б - 0,2 %) в течение 3 ч.: 1 - раствор пероксидазы (С = 0,7 мг/мл), 2 - реакционная смесь, содержащая пероксидазу и перекись водорода
Как видно из рис. 7, даже обычно используемые в научных целях низкие концентрации H2O2 (0,002 %) достаточно быстро разрушают фермент. Увеличение же концентрации перекиси водорода (0,2 %) резко ускоряет этот процесс (через 3 ч. в ЭСП реакционной смеси практически полностью исчезает полоса поглощения Соре, обусловленная наличием порфириновой системы). Однако в присутствии N-оксида 4-стирил-пиридина - субстрата, способного к связыванию с ферментом, но с которым не происходит химическая реакция (рис. 8), - наблюдается совершенно иная картина. После добавления H2O2 к смеси пероксидазы хрена с этим субстратом в ЭСП происходит, как и в предыдущих случаях, батохромный сдвиг полосы поглощения 402 нм ^ 410 нм. Затем постепенно уменьшается интенсивность поглощения при 410 нм, не полно- стью исчезающая даже через несколько дней, свидетельствуя о том, что N-оксид 4-стирилпи-ридина (его спектр со временем практически не изменяется) ингибирует процесс разрушения фермента перекисью водорода. Исследование механизма подобной защиты пероксидаз гетеро-ароматическими N-оксидами является предметом нашей дальнейшей работы.
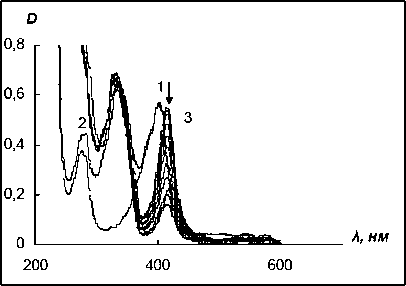
Рис. 8. Изменение ЭСП пероксидазы хрена (С = 0,7 мг/мл) в фосфатном буферном растворе pH 7,3-7,4 в течение 3 ч. в присутствии N-оксида 4-стирилпиридина (С = 1,7 х 10-5 моль/л) и перекиси водорода (С = 0,2 %): 1 - раствор пероксидазы, 2 - раствор пероксидазы и N-оксида 4-стирилпиридина, 3 - реакционная смесь
В дальнейшем опыты по накоплению продуктов реакции между стирильными N-оксидами и пероксидазой хрена выполняли в 50 % этаноле, время от времени добавляя порциями 0,2 % перекись водорода. Контроль за ходом реакции осуществляли методами ТСХ и ВЭЖХ.
Реакции алкилирования и ацилирования N-оксида 4-(4-диметиламиностирилпиридина) осуществляются с участием атома кислорода N-оксидной группы [1], а реакции с ацетиленовыми галогенидами в литературе не описаны. В продолжение исследования нуклеофильных и основных свойств гетероароматических N-оксидов мы решили проверить, по какому центру (центрам) будет происходить реакция пропарги-лирования 4-DPyO 1-бром-2-пропином (бромистым пропаргилом) в ацетонитриле.
Для определения порядка реакции изучаемого процесса по бромистому пропаргилу его концентрацию варьировали (С4-DPyO = 2,17 х 10-5 моль/л) в диапазоне соотношения концентраций от 1:50 000 до 1:20 0000.
Таблица 2
Зависимость наблюдаемой константы скорости реакции (k набл , с-1) пропаргилиро -вания 4-DPyO (С = 2,17 х 10 - 5 моль/л) от концентрации бромистого пропаргила
|
№ п /п |
С(С 3 Н3Вг), моль/л |
k « , с.-1 набл |
k 2 , моль / л • с. |
|
1 |
1,085 |
0,000154 |
0,000142 |
|
2 |
1,63 |
0,000231 |
0,000142 |
|
3 |
2,17 |
0,000292 |
0,000135 |
|
4 |
3,255 |
0,000322 |
|
|
5 |
4,34 |
0,000362 |
lg [С3Н3ВП
б
а
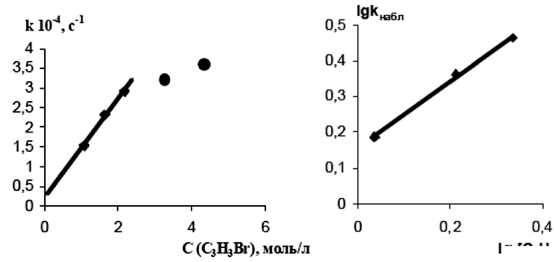
Рис. 9. Зависимость наблюдаемой константы скорости (кнабл) реакции пропаргилирования 4-DPyO (С = 2,17 х 10-5 моль/л) от концентрации бромистого пропаргила в обычных (а) и логарифмических (б) координатах
Зависимость наблюдаемой константы скорости реакции нуклеофильного замещения псевдопервого порядка от концентрации бромистого пропаргила (табл. 2, рис. 9а) имеет сложный характер, однако в диапазоне концентраций бромистого пропаргила 1,09-2,17 моль/л она является линейной, что свидетельствует о первом порядке реакции по бромистому пропаргилу. Это также подтверждается близкой к единице (0,91 ± 0,1) величиной тангенса угла наклона прямой в координатах lg к набл - lg [C3H3Br] (рис. 9б). Среднее значение константы скорости второго порядка к2 из трех значений составляет 0,000140 моль/л • с.
При увеличении соотношения концентраций [C3H3Br]/[4-DPyO] линейный характер наблюдаемой константы от концентрации бромистого пропаргила не сохраняется, что указывает на изменение механизма реакции.
Анализ формальных кинетических данных позволяет предположить, что реакция осуществляется с участием двух реакционных центров стирильного N-оксида. Сначала происходит взаимодействие бромистого пропаргила с N-оксидной группой 4-DPyO, о чем свидетельствует сильный батохромный сдвиг дальневолновой полосы поглощения реакционной смеси от 392 до 495 нм.
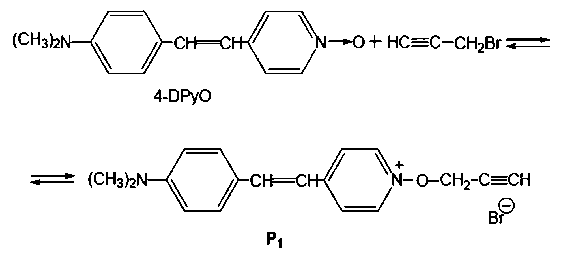
Затем образующийся продукт Р 1 медленно реагирует с избытком пропаргил бромида с участием второго нуклеофильного центра молекулы (атома азота группы N(CH 3 ) 2 ), о чем свидетельствует коротковолновое смещение максимума поглощения дальневолновой полосы поглощения реакционной смеси (495 нм).
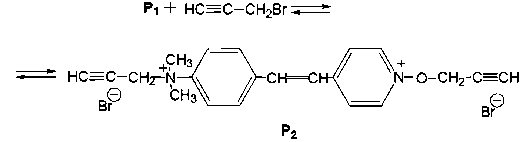
Таким образом, в ацетонитриле первым центром пропаргилирования 4-DPyO является атом кислорода группы N^O, а затем реакция идет по атому азота аминогруппы, что находится в соответствии с литературными данными о последовательности протонирования этих центров в органических растворителях [1], [2]. Ранее в работе [1] нами было показано, что на основании данных электронной спектроскопии можно делать вывод о центре координации 4-DPyO (и других димети-ламиностирильных производных пиридинов и хинолинов) c сильными v-акцепторами в различных водных и органических растворителях. В данном случае взаимодействие нуклеофила осуществляется с образованием продукта реакции, в котором электронная плотность в молекуле смещается в сторону тройной связи, что в зависимости от направления реакции должно приводить к соответствующему смещению в ЭСП дальневолновой полосы поглощения 4-DPyO.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Анилин и его производные очищали, как описано в работе [16], стирильные производные N-оксидов пиридинов и хинолинов синтезировали, как описано в работах [2], [6], [15], [16], их физические константы совпали с литературными. Электронные спектры снимали на приборе СФ 2000-02. Константы устойчивости Zn-ТФП с пиридинами в хлороформе определяли, как описано в работе [3].
Активность препарата пероксидазы хрена фирмы «Диа-М» (удельная активность - 100 ед./мг) имела RZ = 1,4 (RZ - отношение A403/A280, характеризующее процент включения гемина в апо-пероксидазу).
Конечные концентрации веществ в 2 мл пробы (фосфатный буфер, pH 7,2) составляли: 10-5 М гетероароматического N-оксида, 0,002 % Н 2 О 2 и 0,03 мкг/мл пероксидазы хрена.
Активность пероксидазы в тканях растений определяли спектрофотометрически (спектрофотометр СФ-2000, Россия). Основными объектами исследования являлись деревья карельской березы повислой (Betula. pendula var. carelica) с узорчатой структурой древесины. Экспериментальные работы проводили в посадках деревьев, произрастающих в одинаковых почвенно-климатических условиях на Агробиологической станции Карельского научного центра РАН вблизи г. Петрозаводска. Отбор материала проводили в середине июня 2010 года. Из стволовой части отбирали ткани ксилемы, куда входили материнские клетки ксилемы и ксилема текуще- го года. Весь растительный материал сразу замораживали в жидком азоте. Для получения ферментативного препарата ткани ксилемы гомогенизировали при 4 оС в буфере, содержащем 50 мМ Hepes (pH = 7,5), 1 мМ ЭДТА, 1 мМ ЭГТА, 3 мМ DTT, 5 мМ MgCl2, 0,5 мМ PMSF. После 20минутной экстракции гомогенат центрифугировали при 10 000 g в течение 15 мин. (центрифуга К-24, Германия). Осадок троекратно промывали буфером. Осадок и объединенный супернатант диализовали при 4 оС в течение 18-20 ч. против буфера для гомогенизации, разбавленного в 10 раз. Состав реакционной смеси для определения активности пероксидаз в реакции окисления гваякола был следующий: 50 мМ K, Na - фосфатный буфер (рН = 4,9), 4,8 мМ перекись водорода, 21,5 мМ гваякол. Для бензидина: 50 мМ K, Na - фосфатный буфер (рН = 7,8), 2,5 мМ перекись водорода, 33,6 мкМ бензидина. Ферментативную активность пероксидазы определяли: для бензидина по градуировочной кривой (X = 282 нм) выражали как: израсходовано мкмоль бензидина/г сырой ткани; для гваякола - по скорости образования продукта реакции тетрагваякола (с учетом коэффициента экстинкции е 470 нм = 0,0266 мкМ-1 см-1), выражали как: образовалось тетрагваякола/г сырой ткани.
Качественный анализ, а также подтверждение индивидуальности соединений осуществляли методом ВЭЖХ: хроматограф Laboratorni pristroje Praha; колонки Separon SGX C18 (3 х 150 мм); подвижная фаза - ацетонитрил; скорость подачи элюента - 0,4 мл/мин.; детектор ультрафиолетовый LCD 2040; X - 392 нм (4-DPyO) или
268 нм (4-HOOC-PyO, продукт реакции 4-DPyO с пероксидазой хрена, 4-ДМАБК).
Синтез 1-бром-2-пропина (бромистого пропаргила)
В трехгорлую колбу, снабженную мешалкой, капельной воронкой, обратным холодильником и охлажденную до -5 ° С льдом с солью, вносили 36,5 г (0,65 моль) перегнанного пропаргилового спирта и 6 мл пиридина. К этой смеси при перемешивании по каплям прибавляли 61,6 г (0,23 моль) PBr3 в 2 мл пиридина. Температуру реакционной смеси постепенно в течение 1 ч. доводили до комнатной, и смесь кипятили 1 ч. на водяной бане. Затем обратный холодильник заменяли на нисходящий и отгоняли бромистый пропаргил в приемник на сухой поташ. Через некоторый промежуток времени бромистый пропаргил перегоняли еще раз. Выход - 43,3 г (56 %), Т - 84 ° С / 760 мм рт. ст., nD20 - 1,4920 [1].
Контроль за ходом реакции пропаргилирова-ния 4-DPyO в ацетонитриле осуществляли по изменению оптической плотности 4-DPyO (392 нм) и продукта (495 нм).
Значения наблюдаемых констант реакции
псевдопервого порядка при исследовании кинетики реакции 4-DPyO с 1-бром-2-пропином рассчитывали по формуле:
k набл = 1t ln
D
-
D
∞
D t- D
где Do, Dt, Dm - соответственно оптические плот-ности0 растворов в начальный момент времени, в момент времени t и по окончании реакции.
Список литературы Взаимодействие цинк-тетрафенилпорфина, бромистого пропаргила и пероксидазы хрена с анилинами
- Андреев В. П. Молекулярные комплексы гетероароматических N-оксидов и ацетиленовых аминов с v-акцепторами как модель исследования нуклеофильности и основности соединений с пространственно доступными реакционными центрами: Дис.... д-ра хим. наук. Петрозаводск, 2007.
- Андреев В. П., Батоцыренова Е. Г., Рыжаков А. В., Родина Л. Л. Процессы внутримолекулярного переноса заряда в ряду стирильных производных N-оксидов пиридина и хинолина//ХГС. 1998. № 8. C. 1093-1102.
- Андреев В. П., Вапиров В. В., Нижник Я. П., Тунина С. Г., Соболев П. С. Комплексообразование цинк-тетрафенилпорфина и реакции нуклеофильного замещения с участием пиридинов и N-оксидов пиридинов//ЖОрХ. 2010. Т. 46. Вып. 10. С. 1556-1563.
- Андреев В. П., Нижник Я. П., Безручко Д. Г., Морозов А. К. Экстракоординация Zn-тетрафенилпорфина с N-оксидами ряда пиридина, хинолина и акридина//ЖОХ. 2005. Т. 75. Вып. 8. С. 1379-1387.
- Андреев В. П., Нижник Я. П., Лебедева Н. Ш. Новая шкала основности/нуклеофильности, основывающаяся на параметрах, характеризующих образование аксиальных (n, v-типа) комплексов Zn-ТФП с лигандами (основаниями/нуклеофилами)//ЖОрХ. 2008. Т. 44. № 6. С. 914-922.
- Андреев В. П., Рыжаков А. В., Теканова С. Г. Стирильные производные N-оксида хинолина//ХГС. 1995. № 4. C. 518-521.
- Андреева В. А. Фермент пероксидаза: участие в защитном механизме растений. М.: Наука, 1988. 128 с.
- Бах А. Н. Труды по химии и биохимии. М.: Изд-во АН СССР, 1950. 648 с.
- Газарян Г. Особенности структуры и механизма действия пероксидаз растений//Успехи биологической химии. 2006. Т. 46. С. 303-322.
- Днепровский А. С., Темникова Т. И. Теоретические основы органической химии: Учебник для вузов. 2-е изд., перераб. Л.: Химия, 1991. 560 с.
- Кирейко А. В., Веселова И. А., Шеховцев а Т. Н. Механизм реакций пероксидазного окисления о-дианизидина, 3,3',5,5'-тетраметилбензидина и о-фенилендиамина в присутствии додецилсульфата натрия//Биоорганическая химия. 2006. Т. 32. № 1. С. 80-86.
- Литвиненко Л. М., Попов А. Ф., Костенко Л. И., Носовская Т. Д. Влияние структуры транс-арил-β-хлорÔ0 TJ/TT2-винилкетонов на скорость реакции с ариламинами//ЖОрХ. 1973. Т. 9. Вып. 3. С. 552-557.
- Николс П. Оксигеназно-пероксидазная теория Баха и Шода и ее современные эквиваленты: изменение и постоянство в научном мышлении на примере нашего понимания роли воды, перекиси и кислорода в функционировании редокс-ферментов//Биохимия. 2007. Т. 72. Вып. 10. С. 1278-1288.
- Рогожин В. В. Пероксидаза как компонент антиоксидантной системы живых организмов. СПб.: ГИОРД, 2004. 240 с.
- Тицкий Г. Д., Туровская М. К. АС № 1599366. СССР. 1990.
- Biggs A. I. The heat of ionization of some substituted anilines//J. Chem. Soc. 1961. P. 2572-2574.
- Boulton P. D., Hall F. M. Substituent effects on the thermodynamic functions of proton dissociation of para-substituted anilinium ions//J. Chem. Soc. (B). 1969. P. 259-263.
- Brewster M. E., Doerge D. R., Huang M. J., Kaminski J. J., Pop E., Bodor N. Application of semiempirical molecular orbital techniques to the study of peroxidase-mediated oxidation of phenols, anilines, sulfi des and thiobenzamides//Tetrahedron. 1991. Vol. 47. P. 7525-7536.
- Brink C. V. d. M., Jager P. I. N-oksiede van die metielpiridiene. I. Die Daarstellung van Stirielpiridiene wet behulp van N-oksialkielpiridinium-Derivate//Tydskr. Natuurwer. 1963. Vol. 3. P. 74.
- Cambridge Structural Database, 2010 [Electronic resource]. Mode of access: http://www.ccdc.cam.ac.uk
- Candeias L. P., Folke s L. K., Wardman P. Factors controlling the substrate specifi city of peroxidasees: kinetics and termodinamics of the reaction of horseradish peroxidase compound I with phenols and indole-3-acetic acids//Biochemistry. 1997. Vol. 36. P. 7081-7085.
- Dunford H. B., Aderian A. J. Hammett ρσ correlation for reactions of horseradish peroxidase compound II with phenols//Arch. Biochem. Biophys. 1986. Vol. 251. P. 536-542.
- Fenoll L. G., Garcia-Molina F., Gilabe r t M. A., Varon R., Garcia-Ruiz P. A., Tudela J., Garcia-Canova s F., Rodriguez-Lopez J. N. Interpretation of the reactivity of peroxidase compound II with phenols and anilines using the Marcus equation//Biol. Chem. 2005. Vol. 386. P. 351-360.
- Folkes L. K., Candeias L. P. Interpretation of the reactivity of peroxidase compounds I and II with phenols by the Marcus equation//FEBS Lett. 1997. Vol. 421. P. 305-308.
- Job D., Dunford H. B. Substituent effect on the oxidation of phenols and aromatic amines by horseradish peroxidase compound I//Eur. J. Biochem. 1976. Vol. 66. P. 607-614.
- Lee B. C., Yoon J. H., Lee C. G., Lee I. Kinetics and mechanism of aminolisis of benzoic anhydrides//J. Phys. Org. Chem. 1994. Vol. 7. P. 273-279.
- Lee I., Sohn S. C., Kang C. H., Oh Y. J. Nucleophilic substitution of benzyl benzenesulphonates with anilines in methanol-acetonitrile mixtures. Part 2.Variation in transition-state structure//Chem. Soc. Perkin. Trans. II. 1986. № 10. P. 1631-1634.
- Rodriguez-Lopez J. N., Gilabe r t M. A., Tudela J., Thorneley R. N. F., Garcia-Canovas F. Reactivity of horseradish peroxidase compound II toward for a two-step mechanism//Biochem. 2000. Vol. 39. P. 13201-13209.
- Sakurada J., Sekiguchi R., Sato K., Hosoya T. Kinetic and molecular orbital studies on the rate of oxidation of monosubstituted phenols and anilines by horseradish peroxidase compound II//Biochem. 1990. Vol. 29. P. 4093-4098.
- VanHa andel M. J. H., Claassens M. M., Van der Hout N., Boersma M. G., Vervoor t J., Riet jens I. M. Differential substrate behaviour of phenol and aniline derivatives during conversion by horseradish peroxidase//Biochim. Biophys. Acta. 1999. Vol. 1435. P. 22-29.
