Ab initio modelling of a bilayer graphene

Free access
Using the electron density functional theory, numerical modelling of bilayer graphene has been performed. The structure and binding energy of the layers depending on the representation of the wave function of the system have been studied: plane waves (VASP package) and atomic-like orbitals (SIESTA package); and choice of approximation for the exchange-correlation (XC) functional. It has been shown that being in the free form the system creates a stable AB structure of bilayer graphene. The calculation of the layer binding energy has shown that the results of modelling performed with different basis for the wave function are consistent when using XC functionals corresponding to each other and considering the correction to the basis set superposition error in the tight binding method. As expected, the generalized gradient approximation (GGA) has shown underestimated values of the interaction energy of graphene layers. Comparison with experimental data has shown that the energy and geometric characteristics of bilayer graphene are best described by the local electron density approximation (LDA). The 2nd and 3rd generation semi-empirical Grimme corrections for GGA have given estimates of the binding energy higher than LDA, but also close to the experimental results.
Bilayer graphene, density functional theory, ab initio modelling, binding energy
Short address: https://sciup.org/147237462
IDR: 147237462 | UDC: 538.9
Первопринципная модель двухслойного графена
С помощью метода функционала электронной плотности проведено численное моделирование двухслойного графена. Исследовалась структура и энергия связи слоев в зависимости от представления волновой функции системы: плоские волны (пакет VASP) и атомноподобные орбитали (пакет SIESTA) и выбора приближения для обменно-корреляционного (XC) функционала. Было показано, что в свободной форме система приходит к устойчивой АВ-структуре двухслойного графена. Расчет энергии связи слоев показал, что результаты моделирования, выполненного с разным базисом для волновой функции, согласуются при использовании соответствующих друг другу XC-функционалов и учете поправки к ошибке суперпозиции базисного набора в методе сильной связи. Приближение обобщенных градиентов (GGA) ожидаемо дало заниженные значения энергии взаимодействия слоев графена. Сравнение с экспериментальными данными показало, что лучше всего энергетические и геометрические характеристики двухслойного графена описывает приближение локальной электронной плотности (LDA). Полуэмпирические поправки Гримме 2-го и 3-го поколения для GGA дали оценки энергии связи выше, чем LDA, но тоже близкие к экспериментальным результатам.
Text of the scientific article Ab initio modelling of a bilayer graphene
Bilayer graphene (BLG) is an intermediate structure between graphene and graphite consisting of only two graphene sheets. It has three configurations: a metastable AA structure obtained experimentally [1–3], a stable and well-studied AB structure (Bernal configuration [4]), and twisted bilayer graphene (tBLG), where two graphene layers are rotated by a small relative angle [5]. The latter structure has promising properties and may be of interest for nanoelectronics [6, 7], though it is difficult to obtain experimentally, and its model is too demanding for computer simulation. The intermediate A(B) structure (between AA and AB configurations), which has a bandgap of about 0,35 eV [3], also has possible applications in electronics and nanoelectronics, for example, as a channel for a field-effect transistor [1–3]. To investigate the BLG electronic properties, using the density functional theory, we needed the geometrical models. Therefore, in this research, we obtained the models of different BLG configurations and studied their structural properties and the interlayer binding energies.
Models and simulation details
For calculations, we utilized the SIESTA software package with an atomic-like basis set [8] and the VASP code with a plane wave basis set [9]. In both packages, the periodic boundary conditions were implemented. For exchange-correlation potential, we used local electron density approximation, LDA (Ceperley-Alder functional, CA [10]), and generalized gradients approximation, GGA (Perdew-Burke-Ernzerhof functional, PBE [11]). To take into account the van der Waals interactions, we utilized the Grimme semi-empirical corrections, DFT-D2 [12] and DFT-D3 [13]. For the SIESTA calculations, pseudopotentials were taken from the FHI database [14] and [15], for the VASP simulations, we used the 2012 version of pseudopotentials, with 2s22p2 electrons for С as valence.
The simulation cell contained 64 carbon atoms (32 for each graphene layer). The optimized translational parameter was 9,86 Å and 9,79 Å for GGA (GGA+D) and LDA calculations, respectively. We chose the partitioning density of the reciprocal space (k-points) and the real space (the MeshCutoff parameter in the SIESTA package and the Accurate tag in the VASP code) so that the precision of the structure's total energy was ~1 meV. For VASP, the cutoff energy of plane waves was taken as 600 eV. Simulation parameters are presented in Table 1. For one graphene layer, we performed both spin-polarized and not spin-polarized calculations. However, the difference between the resulting total energies was less than 1 meV. Therefore, the bilayer systems we modeled without spin polarization to save computer resources. The resulting precision of the interlayer binding energy was about 0,3 meV/atom.
Table 1
Simulation parameters
|
DFT package |
SIESTA |
VASP |
|
XC approximations |
PBE-GGA, CA-LDA, PBE+D2 |
PBE-GGA, CA-LDA, PBE+D2, PBE+D3 |
|
k -points |
19×19×1 |
9×9×1 |
|
Space partitioning (mesh detailing in the real space): MeshCutoff (SIESTA) / PREC (VASP) |
GGA (+D2): 360 Ry; LDA: 230 Ry |
Accurate |
|
Total energy convergence criterion |
10–6 eV |
10–6 eV |
|
Force convergence criterion |
10–4 Ry/Bohr |
10–3 eV/Å |
|
Vacuum layer in the direction perpendicular to graphene, Å |
at least 20 |
at least 20 |
Basis set optimization
We optimized the localized pseudoatomic orbitals of the carbon atom according to the procedure described in [16]. To minimize the computational time, we simulated the graphene primitive unit cell, which contains only 2 carbon atoms. The cell translation parameter was 2,47 Å, the simulation parameters were the same as in Table 1, except for k -points: in this case, we used a 32×32×1 set. Since the unit cell is small, instead of the interatomic distance, we checked the system “pressure” which indicates how far the translational parameter of the simulation cell, and hence the distances between carbon atoms, are from optimal.
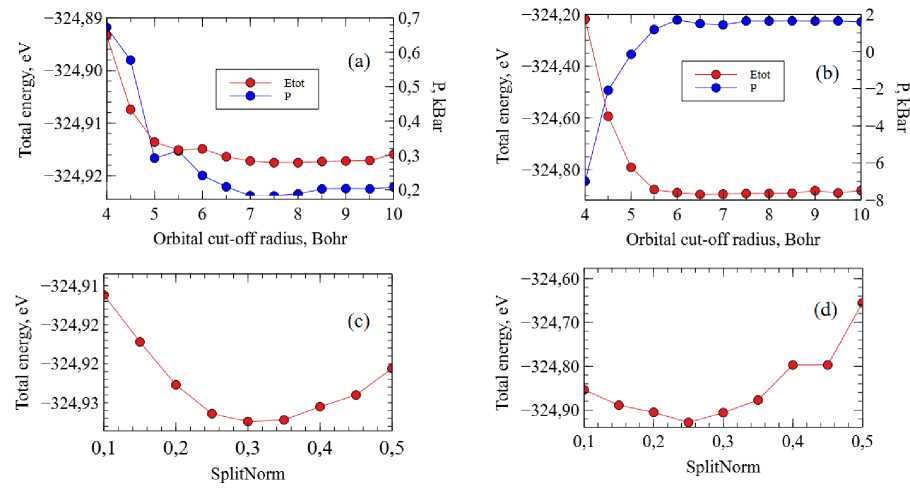
Fig. 1. Optimization of C(2p) and C(2s) orbitals for SIESTA calculations (GGA-PBE). Dependences of the total energy and pressure on the orbital cut-off radius of: (a) C(2s) and (b) C(2p) orbitals; dependence of the system total energy on the parameter SplitNorm for: (c) C(2s) and (d) C(2p) orbital
Figure 1 shows the results of orbital optimization for graphene, simulated with GGA. The used double-ζ basis set could be characterized by two parameters: the orbital cut-off radius, r , and the SplitNorm parameter, which specifies the radius of the modified orbital, r [8]. The optimal parameters were chosen using the criterion of minimal total energy, while the pressure of the simulation cell was additionally monitored. Fig. 1a indicates that both system total energy and pressure were at approxi- Вестник ЮУрГУ. Серия «Математика. Механика. Физика»
2022, том 14, № 2, С. 64–71
mately the same level after the C(2s) orbital cut-off radius reached ~7 Bohr. Since for a larger r more computer resources are needed, we chose r = 7,0 Bohr as optimal. The dependence of the total energy on the SplitNorm parameter had a pronounced minimum at SplitNorm = 0,30, which was chosen as optimal. The optimal parameters of С(2р) orbital were determined similarly (see Fig. 1, b , d ). Optimal basis-set parameters are presented in Table 2.
Table 2
Optimal basis set parameters for C(2p) and C(2s) orbitals, 1 Bohr ≈ 0,529 Å
|
XC approximation |
GGA and GGA+D |
LDA |
||||
|
Orbital |
rcut , Bohr |
SplitNorm |
r Bohr m , |
r Bohr cut , |
SplitNorm |
r Bohr m , |
|
Pseudopotential from [14 |
||||||
|
C(2s) |
7,09 |
0,30 |
2,95 |
7,09 |
0,30 |
2,95 |
|
C(2p) |
6,57 |
0,25 |
3,18 |
6,57 |
0,25 |
3,18 |
|
Pseudopotential from [15 |
||||||
|
C(2s) |
6,57 |
0,25 |
3,11 |
7,09 |
0,30 |
3,03 |
|
C(2p) |
7,09 |
0,25 |
3,22 |
6,57 |
0,25 |
3,18 |
Table 2 shows that the optimal parameters in the case of different XC approximations (GGA and LDA) practically coincide and are close for the chosen pseudopotentials.
Geometry optimization of different BLG configurations
After basis-set optimization, we investigated the structure of bilayer graphene using the SIESTA package. We considered starting configurations with various relative displacements in three directions of one graphene layer to another. The interlayer binding energy was calculated as:
E = E - 2 E - E , (1)
bind 2l 1l CP , where E and E are the total energies of the bilayer system and the isolated layer, respectively, and
E is the Boys–Bernardi correction to the basis set superposition error [17].
Firstly, we shifted the second layer only along the Z-axis (AA structure, Fig. 2a). We tested different initial shifts (from 1 to 15 Å). Though, at ∆z ≥ 10 Å ( ∆z ≥ 5 Å for PBE-GGA calculations) the layers did not interact: the interlayer distance, d , did not change after geometry optimization, and the interaction energy E = 0 eV. All other initial structures moved to a stable state with a certain minimum in- terlayer distance (this distance varied depending on the used XC approximation, see Table 3).
At the next stage, we added a shift along the X -axis. The initial interlayer distance was taken from the stable AA configuration. We considered the initial shifts ∆x in the range of 0,2–0,8 Å (with a step of 0,2 Å). After geometry optimization, we got either the A(B) configuration (Fig. 2, b ) or AB (Fig. 2, c ). In the case of SIESTA DFT-D2 calculations, we observed only the AB . Finally, when a shift along the Y -axis was added (we considered the same initial shifts ∆y as ∆x ), all starting structures transferred to the AB configuration. The obtained values of the interlayer distances and the interlayer binding energies for SIESTA and VASP calculations are presented in Tables 3 and 4, respectively. The values in parentheses (Table 3) show the results obtained with the pseudopotential from [15].
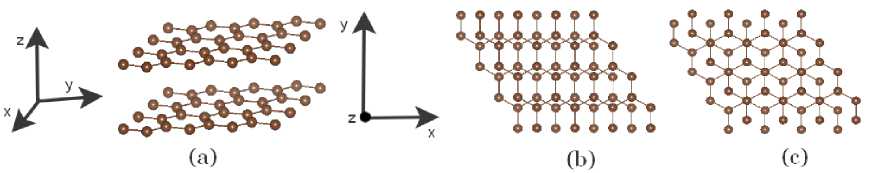
Fig. 2. Atomic structure of the (a) AA system (shift along the Z -axis); (b) A(B ) system (shifts along the X- and Z- axes), and (c) AB system (shifts along the X- , Y- and Z -axes)
Table 3 shows that the GGA calculations predict the weak interaction of the layers or even their repulsion. In previous theoretical studies with various dispersion corrections (including quantum Monte Carlo and DMC methods), the BLG interlayer binding energy for the АА-structure was in the range of 10,4–31,1 meV/atom; for the АВ-structure – in the range of 17,8–70,0 meV/atom [18]. Experimental values for the graphite interlayer binding energy are also very scattered: 0,21–0,37 J/m2 (indirect measurements) and 0,19 J/m2 (direct measurements) [19].
Table 3 The interlayer binding energy, E , and distance, d , calculated using the SIESTA package
|
Structure |
AA |
A(B) |
AB |
||||||
|
XC functional |
PBE |
CA |
PBE+D2 |
PBE |
CA |
PBE+D2 |
PBE |
CA |
PBE+ D2 |
|
d , Å |
3,73 (4,02) |
3,44 (3,45) |
3,38 |
3,53 (3,55) |
3,25 (3,23) |
— |
3,49 (3,48) |
3,23 (3,23) |
3,17 |
|
| d - d exp | , Å |
0,18 (0,47) |
0,11 (0,10) |
0,17 |
– |
– |
– |
0,14 (0,13) |
0,12 (0,12) |
0,18 |
|
— Е bind , meV/atom |
–1,8 (–5,7) |
19,2 (19,1) |
41,6 |
–1,9 (–2,3) |
26,8 (28,3) |
– |
–1,8 (–3,2) |
28,1 (28,3) |
54,1 |
|
– Ebind , J/m |
–0,01 (–0,03) |
0,12 (0,12) |
0,25 |
–0,01 (–0,01) |
0,17 (0,18) |
– |
–0,01 (–0,02) |
0,17 (0,17) |
0,33 |
We chose the empirical graphite interlayer binding energy for comparison because the calculated interlayer E for graphite and BLG are close [20]. Experimentally obtained BLG interlayer distances are 3,55 Å and 3,35 Å for AA and AB configurations, respectively [1]. Therefore, LDA predictions for both the BLG interlayer binding energy and distance are closer to the experimental data than GGA values, although PBE-D2 binding energies are also close to previously obtained experimental and calculated values. Moreover, the difference in the calculation results obtained for considered pseudopotentials was within the calculation error.
Table 4 The interlayer binding energy, E , and distance, d , calculated using the VASP package
|
Structure |
AA |
A(B) |
AB |
|||||||||
|
XC functional |
PBE |
CA |
D2 |
D3 |
PBE |
CA |
D2 |
D3 |
PBE |
CA |
D2 |
D3 |
|
d , Å |
4,50 |
3,60 |
3,52 |
3,71 |
4,39 |
3,36 |
3,29 |
3,55 |
4,38 |
3,32 |
3,25 |
3,53 |
|
| d - d exp | , Å |
0,95 |
0,05 |
0,03 |
0,16 |
– |
– |
– |
– |
1,03 |
0,03 |
0,10 |
0,18 |
|
— Е bind , meV/atom |
1,1 |
14,9 |
38,7 |
39,1 |
1,3 |
22,9 |
48,8 |
43,5 |
1,3 |
24,5 |
50,6 |
44,1 |
|
– Ebind , J/m2 |
0,01 |
0,09 |
0,24 |
0,24 |
0,01 |
0,14 |
0,30 |
0,26 |
0,01 |
0,15 |
0,31 |
0,27 |
The VASP simulations (Table 4) also show that the optimal approximation for studying bilayer graphene is LDA. DFT-D3 corrections tend to predict weaker interlayer binding and larger interlayer distances than DFT-D2. The used simulation packages gave similar results: the difference in binding energies while using the same XC approximations was only 3–4 meV/atom. We observed the smallest difference in the interlayer distance predictions for LDA and DFT+D2 approximations (it did not exceed 0,16 Å, which corresponds to ~4 % difference). The maximum distance variations were observed for GGA-simulated AB configuration: the VASP results exceeded the SIESTA values by 0,9 Å (which corresponds to more than 25 % difference).
Conclusion
In this work, we obtained the optimized model of bilayer graphene and studied its structural and energetic features using the electron density functional method implemented in the VASP and SIESTA packages. For the SIESTA calculations, we optimized the parameters of localized pseudoatomic orbitals for carbon. After the geometry optimization of BLG, we observed a stable Bernal structure ( AB configuration) with a minimum total energy and two metastable structures: AA and A(B) configurations.
The comparison of BLG interlayer binding energies and distances showed that the results obtained by different packages (SIESTA and VASP) were consistent after the simulation parameters optimization (including basis sets) and taking into account the basis set superposition error. Finally, the best agreement with the available experimental data was obtained for the local electron density approximation. Grimme corrections gave the interlayer binding energy close to some experimental results for graphite, too, but its geometrical predictions for the most BLG configurations were less accurate than LDA values. GGA is not suitable for BLG simulation since its application resulted in non-interacting or repulsive layers.
The reported study utilized the supercomputer resources of South Ural State University [21] .
References Ab initio modelling of a bilayer graphene
- Lee J.-K., Lee S.-Ch., Ahn J.-P., Kima S.-C., Wilson J.I.B., John P. The growth of A graphite on (111) diamond. The Journal of Chemical Physics, 2008, Vol. 129, Iss. 23, p. 234709. DOI: 10.1063/1.2975333
- Liu Z., Suenaga K., Harris P.J.F., Iijima S. Open and Closed Edges of Graphene Layers. Physical Review Letters, 2009, Vol. 102, Iss. 1, p. 015501. DOI: 10.1103/physrevlett.102.015501
- Lee, J.K., Kim, J.-G., Hembram, K., Kim, Y., Min, B.-K., Park, Y., Lee, J.-K., Moon, D., Lee, W., Lee, S.-G., John, P. The Nature of Metastable AA' Graphite: Low Dimensional Nano- and Single-Crystalline Forms. Scientific Reports, 2016, Vol. 6, p. 39624. D0I:10.1038/srep39624
- McCann E., Koshino M. The electronic properties of bilayer graphene. Reports on Progress in Physics, 2013, Vol. 76, no. 5, p. 056503 (28pp). DOI: 10.1088/0034-4885/76/5/056503
- Rozhkov A.V., Sboychakov A.O., Rakhmanov A.L., Franco Nori. Electronic properties of graphene-based bilayer systems. Physics Reports, 2016, Vol. 648, pp. 1-104. DOI: 10.1016/j.physrep.2016.07.003
- Dai S., Xiang Y., Srolovitz D.J. Twisted Bilayer Graphene: Moire with a Twist. Nano Letters, 2016, Vol. 16, Iss. 9, pp. 5923-5927. DOI: 10.1021/acs.nanolett.6b02870
- Havener R.W., Zhuang H., Brown L., Hennig R.G., Park J. Angle-Resolved Raman Imaging of Interlayer Rotations and Interactions in Twisted Bilayer Graphene. Nano Letters, 2012, Vol. 12, Iss. 6, pp. 3162-3167. DOI: 10.1021/nl301137k
- Soler J.M., Artacho E., Gale J.D., García A., Junquera J., Ordejón P., Sánchez-Portal D. The SIESTA method for ab initio order-N materials simulation. Journal of Physics: Condensed Matter, 2002, Vol. 14, no. 11, pp. 2745-2779. DOI: 10.1088/0953-8984/14/11/302
- Kresse G. F.J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Physical Review B, 1996, Vol. 54, Iss. 16, pp. 11169-11186. DOI: 10.1103/PhysRevB.54.11169
- Ceperley D.M., Alder B.J. Ground State of the Electron Gas by a Stochastic Method. Physical Review Letters, 1980, Vol. 45, Iss. 7, pp. 566-569. DOI: 10.1103/PhysRevLett.45.566
- Perdew J.P., Burke K., Ernzerhof M. Generalized Gradient Approximation Made Simple. Physical Review Letters, 1996, Vol. 77, no. 18, pp. 3865-3868. DOI: 10.1103/PhysRevLett.78.1396
- Grimme S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. Journal of computational chemistry, 2006, Vol. 27, Iss. 15. pp. 1787-1799. DOI: 10.1002/jcc.20495
- Grimme S., Antony J., Ehrlich S., Krieg H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys., 2010, Vol. 132, no. 15, p. 154104. DOI: 10.1063/1.3382344
- Abinit's Fritz-Haber-Institute (FHI) pseudo database. URL: https://departments.icmab.es/ leem/SIESTA_MATERIAL/Databases/Pseudopotentials/periodictable-intro.html.
- P. Rivero, V.M. Garcia-Suarez, D. Pereniguez et al.. Systematic pseudopotentials from reference eigenvalue sets for DFT calculations: Pseudopotential files. Data in Brief, 2015, Vol. 3, pp. 21-23. DOI: 10.1016/j .dib.2014.12.005
- Anikina E.V., Beskachko V.P. Optimizatsiya parametrov bazisnogo nabora dlya modelirovaniya adsorbtsii vodoroda na uglerodnykh metananotrubkakh v pakete SIESTA (Optimization of the parameters of the basic set for modeling hydrogen adsorption on carbon nanotubes in the SIESTA package). Materialy devyatoy nauchnoy konferentsii aspirantov i doktorantov, Chelyabinsk. 2017 (Proc. Ninth Scientific Conference of Postgraduates and Doctoral Students, Chelyabinsk, 2017), pp. 126-134.
- Boys S.F., Bernardi F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Molecular Physics, 1970, Vol. 19, Iss. 4, pp. 553-566. DOI: 10.1080/00268977000101561
- Mostaani E., Drummond N.D. Fal'ko V.-I. Quantum Monte Carlo calculation of the binding energy of bilayer graphene. Physical Review Letters, 2015, Vol. 115, Iss. 11, pp. 115501. DOI: 10.1103/physrevlett.115.115501
- Liu Z., Liu J.Z., Cheng Y., Li Z., Wang L., Zheng Q. Interlayer binding energy of graphite: a mesoscopic determination from deformation. Physical Review B., 2012, Vol. 85, Iss. 20, p. 205418. DOI: 10.1103/physrevb.85.205418
- Lebegue S., Harl J., Gould T., Angyan J.G., Kresse G., Dobson J.F. Cohesive properties and asymptotics of the dispersion interaction in graphite by the random phase approximation. Physical Review Letters, 2010, Vol. 105, Iss. 19, p. 196401. DOI: 10.1103/physrevlett.105.196401
- Kostenetskiy P., Semenikhina P. SUSU Supercomputer Resources for Industry and fundamental Science. 2018 Global Smart Industry Conference (GloSIC), 2018, pp. 1-7. DOI: 10.1109/GloSIC.2018.8570068
