AB INITIO расчет поверхности потенциальной энергии водородной связи димера воды

Автор: Лебедев Н.Г.
Журнал: Математическая физика и компьютерное моделирование @mpcm-jvolsu
Рубрика: Химическая физика
Статья в выпуске: 1 (14), 2011 года.
Бесплатный доступ
Проведен квантово-химический неэмпирический расчет профиля поверхности потенци- альной энергии водородной связи молекулы димера воды, находящейся в основном и возбуж- денных состояниях, с помощью метода Хартри - Фока с использованием базисных наборов STO-3G, 6-31G*, 6-31++G**. Показано, что во всех выбранных методах ab initio существует минимум потенциала, соответствующий основному состоянию молекулы димера воды. Пока- зано, что для димера в возбужденном состоянии существует два минимума потенциала водо- родной связи, разделенных переходным состоянием.
Димер воды, квантовая химия, водородная связь
Короткий адрес: https://sciup.org/14968661
IDR: 14968661 | УДК: 54.12
AB INITIO calculations of the potential energy surface of the hyrogen binding of water dimer
The quantum chemical calculation of the potential energy surface profile of the hydrogen bond of the water dimer has carried out by ab initio Hartree - Fock methods with STO-3G, 6-31G*, 6- 31++G** basics. It has shown there is the potential minimum corresponding to the ground state of the water dimer in all ab initio methods. It has shown there are two potential minima of the hydrogen bond of the water dimer separated by the transition.
Текст научной статьи AB INITIO расчет поверхности потенциальной энергии водородной связи димера воды
Рентгеноструктурные исследования жидкостей показали, что в них существует некоторый порядок, заключающийся в том, что ближайшее окружение каждой молекулы напоминает, хотя и более рыхлую и подвижную, упаковку в кристаллах. Таким образом, возникает представление о ближнем структурном порядке в жидкостях. Такое понимание структуры справедливо даже для газов, которые (особенно плотные газы) тоже могут характеризоваться неким ближним порядком [1; 5; 7].
В молекуле воды оба водородных атома расположены по одну сторону от атома кислорода, поэтому электрические заряды в ней пространственно рассредоточиваются. Молекула воды полярная, что является причиной особого взаимодействия между разными ее молекулами. Атомы водорода в молекуле воды, имея частичный положительный заряд, взаимодействуют с электронами атомов кислорода соседних молекул. Водородная связь объединяет молекулы воды в своеобразные полимеры пространственного строения. В водяном паре присутствует около 1 % димеров воды. Расстояние между атомами кислорода – 0,3 нм [там же].
На рисунке 1 изображена линейная конфигурация двух взаимодействующих молекул воды в газовой фазе.
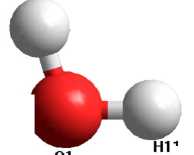
Рис. 1. Структура димера воды
Равновесное расстояние О-Н водородной связи равно 1,78 Е, а энергия разрыва водородной связи составляет примерно 5,5 ккал/моль–1, что на порядок меньше энергии ковалентных связей и на порядок больше энергии дисперсионных взаимодействий [1; 5; 7].
Потенциал водородной связи димера воды в основном состоянии
Для изучения особенностей геометрического и электронного строения молекулы димера воды необходим расчет потенциала водородной связи, благодаря которой молекула существует в устойчивом состоянии.
Потенциальная кривая E ( x ) какой-либо реакции ( x – координата реакции – известная или предполагаемая) может быть рассчитана последовательным вычислением полной энергии E реагирующей системы как функции геометрических параметров вдоль координаты реакции (см. рис. 2). Теплота реакции Q вычисляется как разность энергий основных состояний продуктов реакции E 0’ и реагентов E 0. Энергия активации прямой реакции Ea вычисляется как разность энергий основного состояния активированного комплекса (переходного состояния) E 0 ≠ и реагентов E 0. Энергия активации Ea ’ обратной реакции – разность энергий основного состояния активированного комплекса и продуктов реакции E 0’.
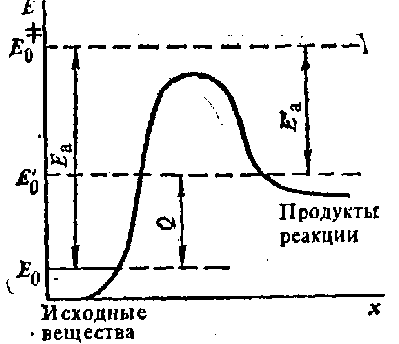
Рис. 2. Потенциальная кривая E ( x ) химической реакции: x – координата реакции – известная или полагаемая [8]
Потенциал водородной связи молекулы димера воды рассчитывался с помощью неэмпирических методов квантовой химии – метода Хартри – Фока – с использованием базисных наборов STO-3G, 6-31G**, 6-31++G** [2; 6].
Для построения профиля поверхности потенциальной энергии E ( x ) в качестве координаты реакции x использовалось межатомное расстояние R (O1-H11) (см. рис. 1). Расчет полной энергии E ( x ) молекулы димера проводился путем пошагового изменения координаты реакции x = R (O1-H11) в пределах от 0,5 до 2,9 Е с шагом ∆ x 0,1 Е. Профили поверхностей потенциальной энергии водородной связи показаны на рисунках 3 и 4 [3; 4].
Как следует из рисунков, во всех выбранных методах ab initio существует минимум потенциала, соответствующий основному состоянию молекулы димера воды. Второй минимум, соответствующий состоянию, когда атом Н11 оказывается связанным с атомом кислорода O2, на кривых отсутствует. В методах 6-31G**, 6-31++G**, включающих диффузные и поляризационные базисные наборы, наблюдается своеобразное «плато» на расстоянии Н11-O2, равном в среднем 0,95 Е. Поиск минимума энергии в данной точке приводит к несходимости процесса минимизации функционала энергии.
Как показали результаты квантово-химических неэмпирических расчетов, поиск переходного состояния, которому соответствует максимум на потенциальной кривой, приводит к раз- рушению системы. Образуется комплекс HO...H3O, в котором атомы кислорода разделены на расстояние свыше 6 Е.
STO-3G
0.0 0.5 1.0 1.52.0
-
-93000 I------------------1-------------------1-------------------11
0.0 0.5 1.0 1.5 2.02.5
6-31G**
-
-95000 I---------------1---------------1---------------1---------------11
2 -93500


-95100
-95200
-95300
-95400

-94500
Координата реакции x=R(O-H), А
-95500
Координата реакции x=R(O-H), A
а б
Рис. 3. Профиль поверхности потенциальной энергии водородной связи димера воды, рассчитанный методом STO-3G ( а ) и 6-31G** ( б )
6-31++G**
0.0 0.5 1.0 1.5 2.0 2.5
-95000 I1111------------------1

-95500
Координата реакции x=R(O-H), A
Рис. 4. Профиль поверхности потенциальной энергии водородной связи димера воды, рассчитанный методом 6-31++G**
Потенциал водородной связи димера воды в возбужденном состоянии
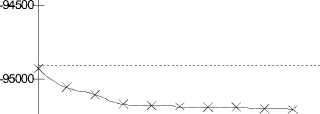
Проведен расчет профиля поверхности потенциальной энергии E ( x ) водородной связи положительно заряженной молекулы димера воды путем пошагового изменения координаты реакции x = R (O 1 -H 11 ) в пределах от 0,5 до 2,4 Е с шагом ∆ x = 0,1 Е. Профили поверхностей потенциальной энергии водородной связи показаны на рисунках 5 и 6 [3; 4].
Как следует из результатов расчета, положительно заряженный димер воды имеет устойчивое геометрическое строение комплекса HO...H-OH2. Как следует из рисунков, во всех выбранных методах ab initio существует минимум потенциала, соответствующий основному состоянию положительно заряженной молекулы димера воды. Поиск второго минимума энергии приводит к несходимости процесса минимизации функционала энергии.
Как показали результаты квантово-химических неэмпирических расчетов, поиск переходного состояния, которому соответствует максимум на потенциальной кривой, приводит к разрушению системы.
Введение в базис диффузных и поляризационных орбиталей для атомов кислорода и водорода в методах 6-31G** и 6-31++G** не приводит к каким-либо принципиальным отличиям ре- зультатов по сравнению с методом STO-3G. Качественно потенциал водородной связи положительно заряженного димера не изменился.
STO-3G
6-31G**
0.6 0.8 1.0 1.2 1.4 1.6
-93000 I--------------1--------------1---------------1--------------1--------------1
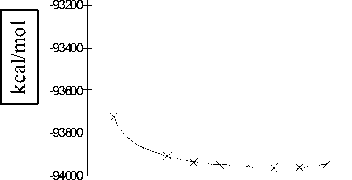
Координата реакции x=R(O-H), A
0.6 0.8 1.0 1.2 1.4 1.6
-94000 I-----------------11111
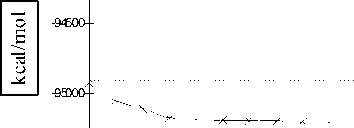
-95500
Координата реакции x=R(O-H), A
а
б
Рис. 5. Профиль поверхности потенциальной энергии водородной связи положительно заряженного димера, рассчитанный методом STO-3G ( а ) и 6-31G** ( б )
6-31++G**
0.6 0.8 1.0 1.2 1.4 1.6
-94000 ,------------------1------------------1------------------1------------------1------------------1

-95500
Координата реакции x=R(O-H), A
Рис. 6. Профиль поверхности потенциальной энергии водородной связи положительно заряженного димера, рассчитанный методом 6-31++G**
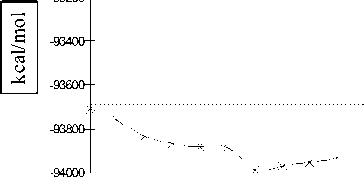
Проведен расчет профиля поверхности потенциальной энергии E ( x ) водородной связи отрицательно заряженной молекулы димера воды путем пошагового изменения координаты реакции x = R (O1-H11) в пределах от 0,5 до 2,4 Е с шагом ∆ x = 0,1 Е. Профили поверхности потенциальной энергии водородной связи показаны на рисунке 7.
Как следует из результатов расчета, отрицательно заряженный димер воды имеет устойчивое геометрическое строение комплекса H2O...OH...H. Во всех выбранных методах ab initio существует только один минимум потенциала, соответствующий основному состоянию положительно заряженной молекулы димера воды. Второй минимум и переходное состояние в потенциале водородной связи отсутствуют.
Как и в предыдущем случае, введение в базис диффузных и поляризационных орбиталей для атомов кислорода и водорода в методе 6-31G** не приводит к каким-либо принципиальным отличиям результатов по сравнению с методом STO-3G. Качественно потенциал водородной связи отрицательно заряженного димера не изменился.
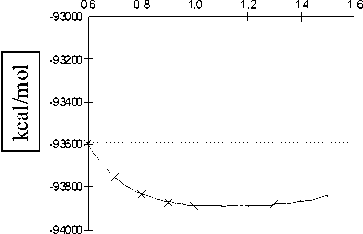
Проведен расчет профиля поверхности потенциальной энергии E(x) водородной связи молекулы димера воды, находящейся в возбужденном состоянии с мультиплетностью χ = 3, путем поша- гового изменения координаты реакции x = R(O1-H11) в пределах от 0,5 до 2,4 Е с шагом ∆x = 0,1 Е.
Потенциалы водородной связи возбужденного димера показаны на рисунке 8.
STO-3G
6-31G**
Координата реакции x=R(O-H), A а
Рис. 7. Профиль поверхности потенциальной энергии водородной связи отрицательно заряженного димера, рассчитанный методом STO-3G ( а ) и 6-31G** ( б )
0.6 0.8 1.0 1.2 1.4 1.6
-95000 I---------------1---------------1---------------1---------------1---------------1
-95100 +

Координата реакции x=R(O-H), A
б
STO-3G
6-31G**
0.6 0.8 1.0 1.2 1.4 1.6
-93000 I-----------------1------------------1------------------1------------------1------------------1
0.0 0.5 1.0 1.5 2.0 2.5
-95000 I------------------1------------------1------------------1------------------1------------------1

-95400
Координата реакции x=R(O-H), A
а
Координата реакции x=R(O-H), A б
Рис. 8. Профиль поверхности потенциальной энергии водородной связи молекулы димера воды в возбужденном состоянии с мультиплетностью χ = 3, рассчитанный методом STO-3G ( а ) и 6-31G** ( б )
Как следует из результатов проведенных квантово-химических расчетов, отрицательно заряженный димер воды имеет устойчивое геометрическое строение комплекса H2O...OH с отрывом одного атома водорода. Существует два минимума потенциала на расстояниях 0,9 и 1,7 Е. В потенциале водородной связи также существует переходное состояние с максимумом энергии на расстоянии 1,6 Е.
Введение в базис диффузных и поляризационных орбиталей для атомов кислорода и водорода в методе 6-31G** приводит к качественному и количественному отличию результатов по сравнению с методом STO-3G. На потенциале водородной связи наблюдается максимум и второй минимум.
Заключение
-
1. Проведен квантово-химический ab initio расчет профиля поверхности потенциальной энергии водородной связи молекулы димера воды, находящейся в основном состоянии, с помо-
- щью метода Хартри – Фока с использованием базисных наборов STO-3G, 6-31G*, 6-31++G**. Показано, что во всех выбранных методах ab initio существует минимум потенциала, соответствующий основному состоянию молекулы димера воды. Второй минимум на кривой отсутствует.
-
2. Проведен квантово-химический ab initio расчет профиля поверхности потенциальной энергии водородной связи положительно и отрицательно заряженной молекулы димера воды с помощью метода Хартри – Фока с использованием базисных наборов STO-3G, 6-31G*, 6-31++G**. Показано, что во всех выбранных методах существует только один минимум потенциала. Переходное состояние отсутствует.
-
3. Проведен квантово-химический ab initio расчет профиля поверхности потенциальной энергии водородной связи молекулы димера воды, находящейся в возбужденном состоянии с мультиплетностью χ = 3, с помощью метода Хартри – Фока с использованием базисных наборов STO-3G, 6-31G*. Показано, что существует два минимума потенциала водородной связи, разделенных переходным состоянием.
Список литературы AB INITIO расчет поверхности потенциальной энергии водородной связи димера воды
- Зацепина, Г. Н. Физические свойства и структура воды/Г. Н. Зацепина. -М.: Изд-во МГУ, 1987. -171 с.
- Минкин, В. И. Теория строения молекул/В. И. Минкин, Б. Я. Симкин, Р. М. Миняев. -Ростов н/Д: Феникс, 1997. -560 с.
- Пономарева, Ю. В. Квантово-химическое исследование электронного строения и структуры кластеров воды: вып. квалификац. работа бакалавра/Ю. В. Пономарева. -Волгоград: Изд-во ВолГУ, 2008. -65 с.
- Пономарева, Ю. В. Электронное строение и физико-химические свойства планарных кластеров воды: дис. магистра/Ю. В. Пономарева. -Волгоград: Изд-во ВолГУ, 2010. -97 с.
- Саркисов, Г. Н. Структурные модели воды/Г. Н. Саркисов//УФН. -2006. -Т. 176, № 8. -С. 833-845.
- Степанов, Н. Ф. Квантовая механика и квантовая химия/Н. Ф. Степанов. -М.: Мир, 2001. -519 с.
- Стехин, А. А. Структурированная вода: нелинейные эффекты/А. А. Стехин, Г. В. Яковлева. -М.: Изд-во ЛКИ, 2008. -320 с.
- Эмануэль, Н. М. Курс химической кинетики/Н. М. Эмануэль, Д. Г. Кнорре. -М.: Высш. шк., 1984. -463 с.
