Analysis of Tandem Repeat Patterns in Nlrc4 using a Motif Model

Author: Sim-Hui Tee
Journal: International Journal of Information Technology and Computer Science(IJITCS) @ijitcs
Article in issue: 1 Vol. 5, 2013.
Free access
Exponential accumulation of biological data requires computer scientists and bioinformaticians to improve the efficiency of computer algorithms and databases. The recent advancement of computational tools has boosted the processing capacity of enormous volume of genetic data. This research applied a computational approach to analyze the tandem repeat patterns in Nlrc4 gene. Because the protein product of Nlrc4 gene is important in detecting pathogen and triggering subsequent immune responses, the results of this genetic analysis is essential for the understanding of the genetic characteristics of Nlrc4. The study on the distribution of tandem repeats may provide insights for drug design catered for the Nlrc4-implicated diseases.
Bioinformatics, Algorithm, Database, Tandem Repeat, Nlrc4, Gene
Short address: https://sciup.org/15011803
IDR: 15011803
Text of the scientific article Analysis of Tandem Repeat Patterns in Nlrc4 using a Motif Model
Published Online December 2012 in MECS DOI: 10.5815/ijitcs.2013.01.06
The advancement of computational tools such as databases [1-5], modelling [6-8], decision support systems [9], simulation [10-12], visualization techniques [13-16], and web servers [17-20] have revolutionized the field of bioinformatics and molecular biology with the breakthrough in data processing and analysis. These computational tools received wide application in various specialized disciplines in molecular biology, including systems biology [21-22], genome analysis [23-24], population genetics [25], structural bioinformatics [26-27], and phylogenetics [28-29]. Various biological data have been processed efficiently and accurately with the powerful computational tools at hand. Bioinformaticians and computer scientists have employed these tools to study protein functions [30-34], gene expression [35-38], regulatory gene identification [39-41], and the evolution of gene [42-43].
This research adopts a computational analysis to identify the tandem repeat patterns in Nlrc4 gene. Nlrc4 gene encodes for NLRC4 protein, which is a caspase recruitment domain (CARD) containing, NODlike receptor (NLR) family member that plays a crucial role in innate sensing of pathogens. It is one of the known genes that involves in the formation of inflammasome, which is a molecular complex that mediates the cytosolic pathogen detection and signal cascading [44,46-47]. Notably, the activation of NLRC4 inflammasome by Salmonella and Legionella requires flagellin and functional type III or IV secretion systems [45,49]. Other route of activation is possible as S. flexneri, which is a microorganism lacking of flagellin, can be sensed by NLRC4 [51]. An NLRC4 inflammasome is self-oligomerized and formed by domain interactions between its CARD domain with the CARD domain of pro-caspase-1 [48]. This interaction leads to the cleavage of pro-caspase-1 into a form of activated caspase-1, which causes rapid cell death [50]. Cell death caused by caspase-1 activation will stimulate immune response to eliminate the dead cells.
This research aims to analyze the tandem repeat patterns in Nlrc4 gene which encodes for NLRC4 inflammasome. Tandem repeats are unstable genomic elements which are repeated several times. For instance, the nucleotide sequence CGCGCGCG is a tandem repeat of nucleotides cytosine (C) and guanine (G) for four times; and sequence AGTAGTAGT is a tandem repeat of adenine (A), guanine (G) and thymine (T) for three times. Tandem repeats are ubiquitous in the genome of organisms across species, and the mutations in the regions of tandem repeat have phenotypic consequences [52], such as diseases. In inflammasome aggregates, tandem repeats can be found in the leucine-rich repeat (LRR) region. LRR is also responsible for other non-immunity role, such as the development and functions of neural circuits [53]. The emergence of tandem repeat drives the evolution of organisms, and multiple copies of gene in the form of tandem repeat allow cells to survive under growth-limiting milieu [54]. Hence, analysis of tandem repeats may provide insights in the pathogenesis and the evolution of an organism.
This paper elaborates the methods used in the Section 2. A motif model is provided in this section. Results of tandem repeat patterns are provided in Section 3, with the analysis of the distribution patterns and the signaling pathways. Finally, Section 4 concludes the findings of the tandem repeat patterns of Nlrc4 gene.
-
II. Methods
Sequence data of Nlrc4 gene of human was retrieved from NCBI GenBank. The UCSC genome browser [55]
was used to visualize the locus of Nlrc4 gene on the chromosome. KEGG database [56] was used to extract and match the regulatory and metabolic pathways of NLR signaling pathway. We used mreps algorithm [57] to compute the statistically expected repeats. A motif
M model was used to identify the motif 0 from the nonmotif 0 [58]. The non-motif is the background motif which does not display the pattern of tandem repeat.
The motif model [58] is depicted in (1) below:
|
"oWM... |
0M " _, w |
||
|
B MM “ a 1,o “ a 1,1 “ a 1,2 |
M • • “a 1, w |
||
|
0 = { 0 B , 0 M } = |
B MM “ a 2,0 “ a 2,1 “ a 2,2 |
M . . “ a 2, w |
|
|
e B ,o “ 0M, 1 “ M 2 ■ |
'" M • ' “aj , w |
(1) |
The subsequence of length W at position j in a sequence i is denoted by Sij . Let a be the symbol that
MB occurs at a position k of either 0 or 0 ; let the position k be 1 — k — W, and L be a non-empty set for the length of nucleotide sequence. The conditional probabilities that Sij is found using the motif model are [58]:
WL
P M ( Sij ) =
П П “ M ) I ( S jk - 1 = a )
k = 1 a = 1
the conditional probabilities that Sij is found using the non-motif model are computed as [58]:
Let X be the prior probability of motif occurrence in the gene sequences of Nlrc4 gene. The motif occurrence probability Z at position j in sequence i is derived from the formula below [58]:
WmS"
Z. = ( X P „ S i ) + (1 - X ) P b S“ (4)
Furthermore, a genuine motif will fulfill the following condition [58]:
log ( P „( S »)/ P b ( S j ) ) > log[(1 - X ) X ] (5)
In gene analysis, we need to take the phenomenon of bogus identification of gene motif into account. Such bogus motif shall be discarded. A particular count of motif is taken to be a pseudo-count by [58]:
L pscak = ^ “b,k +1 Pa / b b=1 (6)
Where Pa/b is the BLOSUM substitution probability for amino acid a from the observed amino acid b . Pseudo-count of motif will be discarded in the finalized count.
-
III. Results and Discussion
Nlrc4 is a gene with a length of 3385 base pairs (bp), with the nucleotide composition of A=28.98%, T=25.91%, G=23.40%, and C=21.71%. The gene locus is depicted in Fig. 1.
WL
П П <) I ( '-k - 1 = a )
P b ( S i ) = k = 1 a = 1 (3)
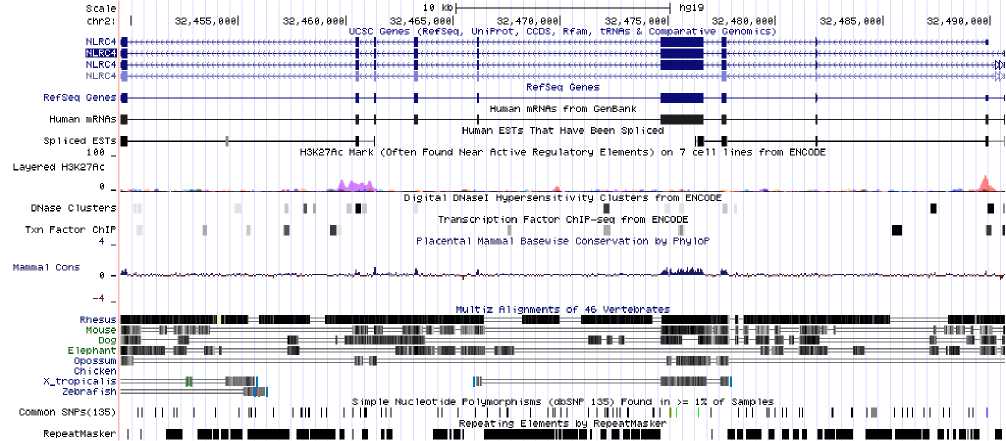
Fig. 1: Nlrc4 on the chromosome
Fig. 1 shows that Nlrc4 is located at chromosome 2. The lower panel of the figure shows that there are extensive repeating elements in the gene region. We identified a particular sequence to be exhibiting a pattern of tandem repeats when a stretch of mononucleotides repeat at least 4 times, and twice for di-, tri-, tetra-, penta-nucleotides. Table 1 illustrates the occurrence and relative frequency of tandem repeat motifs. The relative frequency was computed as per 1k bp.
Table 1: The occurrence and relative frequency of tandem repeat motifs
|
Tandem Repeat Motif |
Occurrence |
Relative Frequncy |
|
Mononucleotide |
53 |
179.41 |
|
Dinucleotide |
111 |
375.74 |
|
Trinucleotide |
33 |
111.71 |
|
Tetranucleotide |
0 |
0 |
|
Pentanucleotide |
0 |
0 |
The results in Table 1 show that the dinucleotide tandem repeat is the most prevalent repeat pattern in Nlrc4 gene. Its relative frequency is double than that of the mononucleotide tandem repeat. We observed that tandem repeats have declined as the order of nucleotide number increases. There are 33 occurrences (relative frequency=111.71) of trinucleotide tandem repeats, whereas none was found in tetranucleotide and pentanucleotide. The absence of tetranucleotide and pentanucleotide tandem repeats can be explained by the small size of Nlrc4 gene.
The distribution of tandem repeats was recorded for each 1kbp of Nlrc4 gene, as shown in Table 2. The numbers in each column indicate the number of occurrence.
Table 2: The distribution of tandem repeats across the Nlrc4 gene region
|
Tandem Repeat Motif |
1-1kbp |
1kbp-2kbp |
2kbp-3385bp |
|
Mononucleotide |
19 |
9 |
25 |
|
Dinucleotide |
34 |
39 |
38 |
|
Trinucleotide |
10 |
8 |
15 |
From Table 2, it was observed that both mononucleotide and trinucleotide tandem repeat exhibit a pattern of reducing occurrence of tandem repeats in the mid-locus region of Nlrc4 gene. However, the reducing number of trinucleotide tandem repeats in the mid-locus region is not statistically significant as the difference from the other regions is not large. As for dinucleotide tandem repeat distribution, we observed an even distribution in all regions of Nlrc4 gene. Besides, we also identified the number of tandem repeat occurrence at the vicinity of 5’ and 3’ region of Nlrc4 gene. The vicinity is defined as 100 bp downstream and upstream of 5’ and 3’ region, respectively. The observed occurrence of tandem repeat was low in 5’ and
3’ vicinity of Nlrc4 gene. Hence, tandem repeats may not have significant impacts on the promoter binding and pathogen sensing in a case where they are mutated in these regions. The results are shown in Table 3.
Table 3: The occurrence of tandem repeat at the vicinity of 5’ and 3’ region of Nlrc4 gene
|
Tandem repeat motif |
5’ vicinity |
3’ vicinity |
|
Mononucleotide |
3 |
2 |
|
Dinucleotide |
3 |
3 |
|
Trinucleotide |
0 |
0 |
We observed a few tandem repeats which occur in an order higher than 4, as given in Table 4.
Table 4: The tandem repeats occur in an order > 4
|
Repeat motif |
Start locus |
End locus |
|
(A) 6 |
178 |
183 |
|
(A) 13 |
3373 |
3385 |
|
(A) 5 |
453 |
457 |
|
(T) 6 |
3063 |
3068 |
|
(T) 6 |
3180 |
3185 |
|
(A) 5 |
1392 |
1396 |
|
(A) 5 |
1887 |
1891 |
|
(A) 5 |
2637 |
2641 |
|
(A) 5 |
2857 |
2861 |
|
(A) 5 |
3018 |
3022 |
|
(A) 5 |
3084 |
3088 |
|
(T) 5 |
541 |
545 |
|
(T) 5 |
655 |
659 |
|
(T) 5 |
910 |
914 |
|
(G) 5 |
1166 |
1170 |
|
(G) 5 |
2127 |
2131 |
The relative frequency of the high order (>4) tandem repeats is only 54.16, implying that for every 1000bp, there are only about 54 bp that are the constituent nucleotide of a higher order tandem repeats. Notice also that all of the high order tandem repeats listed in Table 4 are mononucleotide tandem repeats. The percentage of high order mononucleotide tandem repeat is 30.20% of the total number of mononucleotide tandem repeat occurrence, which is considerably high (for mononucleotide only). However, high order tandem repeats are always scarce in general, as it was also reported by other researcher, such as the similar findings reported by Gur-Arie et al. [59] in their analysis on E. coli .
The NLR signaling pathways of Nlrc4 protein product were identified and matched using KEGG database [56]. These pathways are illustrated in Fig. 2.
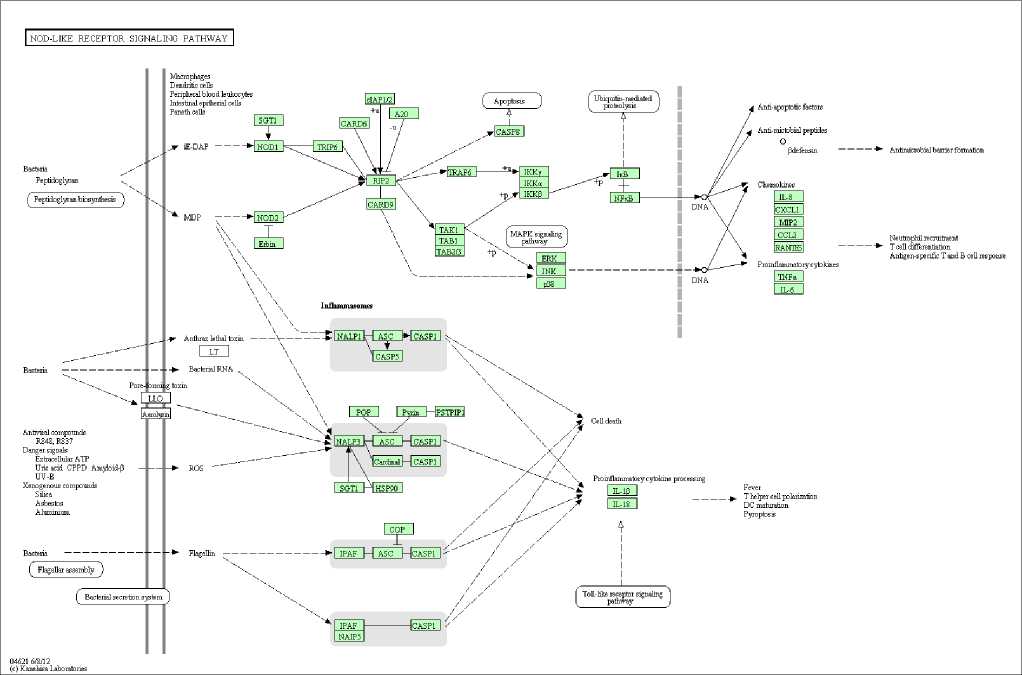
Fig. 2: NLR signaling pathway of NLRC4
Fig. 2 demonstrates that the signaling pathways are originated from bacteria, antiviral chemical compounds, danger signals derived from host cells, and xenogenous compounds such as silica. NOD-like receptor (NLR) which can sense a wide variety of pathogen molecular patterns and danger molecular patterns has four major pathways, which lead to either cell death or the activation of caspase-1 to induce the expression of pro-inflammatory cytokines. The first pathway (the upper panel in Fig. 2) is a signaling pathway that detects the peptidoglycan of bacteria. It is an inflammasome-independent pathway mediated by NOD1 and NOD2 proteins, which culminates in the activation of transcription factor NF- κ B. NF- κ B plays an important role in multiple pathogen sensing mechanisms [60-63], leading to the expression of pro-inflammatory cytokines and chemokines. The second pathway (the middle panel in Fig. 2) demonstrates a cascade of pathways mediated by inflammasome which detects toxins produced by bacteria. This pathway is not totally independent from the first pathway because the inflammasome can be activated by bacterial cell wall component muramyl dipeptide (MDP). This pathway culminates in either cell death or the activation of pro-inflammatory cytokine, which leads to the release of IL-1β and IL-18 to extracellular space. The third pathway is mediated by reactive oxygen species (ROS) that present in cytosol of cells in the condition of cellular stress. The presence of ROS can activate inflammasome, which results in the cleavage of pro-IL-1β to a mature form of IL-1β [64]. The fourth, and the last, pathway (the lowest panel in Fig. 2) is mediated by bacterial flagellin that activates NLRC4 inflammasome. Similar to the second signaling pathway, the cellular outcome of this NLRC4 inflammasome-activated pathway culminates in either cell death or the release of active pro-inflammatory cytokines.
-
IV. Conclusion
Nlrc4 is a gene that encodes for NLRC4 protein (inflammasome) which is implicated in pathogen sensing and the corresponding immune responses. This research applied a computational approach to analyze the tandem repeat patterns in Nlrc4 gene. We found that the dinucleotide tandem repeat is the most prevalent repeat pattern in Nlrc4 gene. In our analysis, the occurrence of tandem repeat was low in 5’ and 3’ vicinity of Nlrc4 gene, implying that the mutated tandem repeats may not have significant impacts on the promoter binding and pathogen sensing. Although as much as 30.20% of mononucleotide tandem repeats occur for more than 4 times consecutively, the high order tandem repeats are always scarce. The signaling pathway demonstrates that the normal expression of Nlrc4 gene is essential for the activation of immune response in the presence of pathogens. Particular attention needs to be paid to the mono-, di-, and trinucleotide tandem repeat mutation in the diagnosis of Nlrc4-implicated diseases. This research provides an insight into the therapeutic strategies and drug designs for Nlrc4-related diseases.
References Analysis of Tandem Repeat Patterns in Nlrc4 using a Motif Model
- S. Philippi, “Light-weight integration of molecular biological databases,” Bioinformatics, Vol. 20, No. 1, pp. 51-57, 2004.
- A. Kiran and P.V. Baranov, “DARNED: a Database of RNa Editing in humans,” Bioinformatics, Vol. 26, No. 14, pp. 1772-1776, 2010.
- Y. Zhang and V.N. Gladyshev, “dbTEU: a protein database of trace element utilization,” Bioinformatics, Vol. 26, No. 5, pp. 700-702, 2010.
- S-Y. Ku and Y-J. Hu, “Structural alphabet motif discovery and a structural motif database,” Computers in Biology and Medicine, Vol. 42, pp. 93-105, 2012.
- D. Vallenet, S. Engelen, D. Mornico, S. Cruveiller, L. Fleury, A. Lajus, Z. Rouy, D. Roche, G. Salvignol, C. Scarpelli, and C. Médigue, “MicroScope: a platform for microbial genome annotation and comparative genomics,” Database, Vol. 2009, bap021, 2009.
- D. Kheffache and O. Ouamerali, “Some hysicochemical properties of the antitumor drug thiotepa and its metabolite tepa as obtained by density functional theory (DFT) calculations,” Journal of Molecular Modeling, Vol. 16, pp. 1383-1390, 2010.
- B. Chen and D.J. Wild, “PubChem BioAssays as a data source for predictive models,” Journal of Molecular Graphics and Modelling, Vol. 28, pp. 420-426, 2010.
- A.P.S. de Moura, R. Retkute, M. Hawkins, and C.A. Nieduszynski, “Mathematical modelling of whole chromosome replication,” Nucleic Acids Research, Vol. 38, No. 17, pp. 5623-5633, 2010.
- E.J. Baede, E. den Bekker, J-W. Boiten, D. Cronin, R. van Gammeren, and J. de Vlieg, “Integrated Project Views: Decision support platform for drug discovery project teams,” Journal of Chemical Information and Modeling, Vol. 52, pp. 1438-1449, 2012.
- L.R. Cooper, D.W. Corne, and M.J.C. Crabbe, “Use of a novel Hill-climbing genetic algorithm in protein folding simulations,” Computational Biology and Chemistry, Vol. 27, pp. 575-580, 2003.
- W. Knight, W. Hill, and J.S. Lodmell, “Ribosome Builder: A software project to simulate the ribosome,” Computational Biology and Chemistry, Vol. 29, pp. 163-174, 2005.
- H.A. Ross, S. Murugan, and W.L.S. Li, “Testing the reliability of genetic methods of species identification via simulation,” Systems Biology, Vol. 57, No. 2, pp. 216-230, 2008.
- M. Brameier and C. Wiuf, “Co-clustering and visualization of gene expression data and gene ontology terms for Saccharomyces cerevisiae using self-organizing maps,” Journal of Biomedical Informatics, Vol. 40, pp. 160-173, 2007.
- J.D. Moore and R.G. Allaby, “TreeMos: a high-throughput phylogenomic approach to find and visualize phylogenetic mosaicism,” Bioinformatics, Vol. 24, No. 5, pp. 717-718, 2008.
- L. Florea, M. McClelland, C. Riemer, S. Schwartz, and W. Miller, “EnteriX 2003: visualization tools for genome alignments of Enterobacteriaceae, ” Nucleic Acids Research, Vol. 31, No. 13, pp. 3527-3532, 2003.
- D. Ristic, R. Kanaar, and C. Wyman, “Visualizing RAD51-mediated joint molecules: implications for recombination mechanism and the effect of sequence heterology,” Nucleic Acids Research, Vol. 39, No. 1, pp. 155-167, 2011.
- A. Alexandridou, N. Dovrolis, G.T. Tsangaris, K. Nikita, and G. Spyrou, “PepServe: a web server for peptide analysis, clustering and visualization,” Nucleic Acids Research, Vol. 39, pp. W381-W384, 2011.
- T. Curk, G. Rot, and B. Zupan, “SNPsyn: detection and exploration of SNP-SNP interactions,” Nucleic Acids Research, Vol. 39, pp. W444-W449, 2011.
- G. Nimrod, M. Schushan, A. Szilágyi, C. Leslie, and N. Ben-Tal, “iDBPs: a web server for the identification of DNA binding proteins,” Bioinformatics, Vol. 26, No. 5, pp. 692-693, 2010.
- J. Oberto, “BAGET: a web server for the effortless retrieval of prokaryotic gene context and sequence,” Bioinformatics, Vol. 24, No. 3, pp. 424-425, 2008.
- S-Y. Wang, T-J. Ho, C-H. Kuo, and Y.J. Tseng, “Chromaligner: a web server for chromatogram alignment,” Bioinformatics, Vol. 26, No. 18, pp. 2338-2339, 2010.
- A. Lachmann, H. Xu, J. Krishnan, S.I. Berger, A.R. Mazloom, and A. Ma’ayan, “ChEA: transcription factor regulation inferred from integrating genome-wide ChIP-X experiments,” Bioinformatics, Vol. 26, No. 19, pp. 2438-2444, 2010.
- S. Ivakhno and S. Tavaré, “CNAnova: a new approach for finding recurrent copy number abnormalities in cancer SNP microarray data,” Bioinformatics, Vol. 26, No. 11, pp. 1395-1402, 2010.
- K. Hanada, K. Akiyama, T. Sakurai, T. Toyoda, K. Shinozaki, and S-H. Shiu, “sORF finder: a program package to identify small open reading frames with high coding potential,” Bioinformatics, Vol. 26, No. 3, pp. 399-400, 2010.
- F.F. Gonzalez-Galarza, S. Christmas, D. Middleton, and A.R. Jones, “Allele frequency net: a database and online repository for immune gene frequencies in worldwide populations,” Nucleic Acids Research, Vol. 39, pp. D913-D919, 2011.
- S. Loriot, F. Cazals, and J. Bernauer, “ESBTL: efficient PDB parser and data structure for the structural and geometric analysis of biological macromolecules,” Bioinformatics, Vol. 26, No. 8, pp. 1127-1128, 2010.
- W. Hugo, F. Song, Z. Aung, S-K. Ng, and W-K. Sung, “SLiM on Diet: finding short linear motifs on domain interaction interfaces in Protein Data Bank,” Bioinformatics, Vol. 26, No. 8, pp. 1036-1042, 2010.
- T. Jombart, F. Balloux, and S. Dray, “adephylo: new tools for investigating the Phylogenetic signal in biological traits,” Bioinformatics, Vol. 26, No. 5, pp. 1907-1909, 2010.
- S.A. Smith and C.W. Dunn, “Phyutility: a phyloinformatics tool for trees, alignments and molecular data,” Bioinformatics, Vol. 24, No. 5, pp. 715-716, 2008.
- M. Deng, K. Zhang, S. Mehta, T. Chen, and F. Sun, “Prediction of protein function using protein-protein interaction data,” Journal of Computational Biology, Vol. 10, No. 6, pp. 947-960, 2003.
- M.A. Erdmann, “Protein similarity from knot theory: geometric convolution and line weavings,” Journal of Computational Biology, Vol. 12, No. 6, pp. 609-637, 2005.
- E. August, K.H. Parker, and M. Barahona, “A dynamical model of lipoprotein metabolism,” Bulletin of Mathematical Biology, Vol. 69, pp. 1233-1254, 2007.
- P.J. Woodroffe, L.J. Bridge, J.R. King, C.Y. Chen, and S.J. Hill, “Modelling of the activation of G-protein coupled receptors: drug free constitutive receptor activity,” Journal of Mathematical Biology, Vol. 60, pp. 313-346, 2010.
- A. Barysenka, A.W.M. Dress, and W. Schubert, “An information theoretic thresholding method for detecting protein colocalizations in stacks of fluorescence images,” Journal of Biotechnology, Vol. 149, pp. 127-131, 2010.
- H. Binder, H. Wirth, and J. Galle, “Gene expression density profiles characterize modes of genomic regulation: Theory and experiment,” Journal of Biotechnology, Vol. 149, pp. 98-114, 2010.
- J.M.G. Vilar, “Accurate prediction of gene expression by integration of DNA sequence statistics with detailed modeling of transcription regulation,” Biophysical Journal, Vol. 99, pp. 2408-2413, 2010.
- X. Xu, Y. Zhao, and R. Simon, “Gene set expression comparison kit for BRB-ArrayTools,” Bioinformatics, Vol. 24, No. 1, pp. 137-139, 2008.
- P. Merz, “Analysis of gene expression profiles: an application of memetic algorithms to the minimum sum-of-squares clustering problem,” BioSystems, Vol. 72, pp. 99-109, 2003.
- M. Hackl, V. Jadhav, T. Jakobi, O. Rupp, K. Brinkrolf, A. Goesmann, A. Pühler, T. Noll, N. Borth, and J.Grillari, “Computational identification of microRNA gene loci and precursor microRNA sequences in CHO cell lines,” Journal of Biotechnology, Vol. 158, pp. 151-155, 2012.
- A. Mathelier and A. Carbone, “MIReNA: finding microRNAs with high accuracy and no learning at genome scale and from deep sequencing data,” Bioinformatics, Vol. 26, No. 18, pp. 2226-2234, 2010.
- W.J. Hsieh and H. Wang, “Human microRNA target identification by RRSM,” Journal of Theoretical Biology, Vol. 286, pp. 79-84, 2011.
- E.S. Allman, M.T. Holder, and J.A. Rhodes, “Estimating trees from filtered data: Identifiability of models for morphological phylogenetics,” Journal of Theoretical Biology, Vol. 263, pp. 108-119, 2010.
- V.V. Lukashov and J. Goudsmit, “Recent evolutionary history of human immunodeficiency virus type 1 subtype B: Reconstruction of epidemic onset based on sequence distances to the common ancestor,” Journal of Molecular Evolution, Vol. 54, pp. 680-691, 2002.
- M.H. Shaw, T. Reimer, Y-G. Kim, and G. Nuñez, “NOD-like receptors (NLRs): bona fide intracellular microbial sensors,” Current Opinion in Immunology, Vol. 20, pp. 337-382, 2008.
- E. Miao, C. Alpuche-Aranda, M. Dors, A. Clark, M. Bader, S. Miller, and A. Aderem, “Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1beta via Ipaf,” Nature Immunology, Vol. 7, pp. 569-575, 2006.
- D.J. Philpott and S.E. Girardin, “Nod-like receptors: sentinels at host membranes,” Current Opinion in Immunology, Vol. 22, pp. 428-434, 2010.
- F. Martinon, A. Mayor, J. Tschopp, “The inflammasomes: guardians of the body,” Annual Review of Immunology, Vol. 27, pp. 229-265, 2009.
- K. Schroder and J. Tschopp, “The inflammasomes,” Cell, Vol. 140, pp. 821-832, 2010.
- L. Franchi, J. Stoolman, T.D. Kanneganti, A. Verma, R. Ramphal, and G. Nuñez, “Critical role for Ipaf in Pseudomonas aeruginosa-induced caspase-1 activation,” European Journal of Immunology, Vol. 37, pp. 3030-3039, 2007.
- S.L. Fink and B.T. Cookson, “Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages,” Cell Microbiology, Vol. 8, pp. 1812-1825, 2006.
- M. Lamkanfi and V. Dixit, “Inflammasomes: guardians of cytosolic sanctity,” Immunological Reviews, Vol. 227, pp. 95-105, 2009.
- R. Gemayel, M.D. Vinces, M. Legendre, and K.J. Verstrepen, “Variable tandem repeats accelerate evolution of coding and regulatory sequences,” Annual Review of Genetics, Vol. 44, pp. 445-477, 2010.
- J. de Wit, W. Hong, L. Luo, and A. Ghosh, “Role of leucine-rich repeat proteins in the development and function of neural circuits,” Annual Review of Cell and Developmental Biology, Vol. 27, pp. 697-729, 2011.
- A.B. Reams and E.L. Neidle, “Selection for gene clustering by tandem duplication,” Annual Review of Microbiology, Vol. 58, pp. 119-142, 2004.
- P.A. Fujita, B. Rhead, A.S. Zweig, A.S. Hinrichs, D. Karolchik, M.S. Cline, M. Goldman, G.P. Barber, H. Clawson, A. Coelho, M. Diekhans, T.R. Dreszer, B.M. Giardine, R.A. Harte, J. Hillman-Jackson, F. Hsu, V. Kirkup, R.M. Kuhn, K. Learned, C.H. Li, L.R. Meyer, A. Pohl, B.J. Raney, K.R. Rosenbloom, K.E. Smith, D. Haussler, and W.J. Kent, “The UCSC Genome Browser database: update 2011”, Nucleic Acids Research, Vol. 39, pp. D876-D882, 2011.
- M. Kanehisa, S. Goto, S. Kawashima, and A. Nakaya, “The KEGG databases at GenomeNet,” Nucleic Acids Research, Vol. 30, pp. 42-46, 2002.
- R. Kolpakov, G. Bana, and G. Kucherov, “mreps: efficient and flexible detection of tandem repeats in DNA,” Nucleic Acids Research, Vol. 31, No. 13, pp. 3672-3678, 2003.
- T. Le, T. Altman, and K. Gardiner, “HIGEDA: a hierarchical gene-set genetics based algorithm for finding subtle motifs in biological sequences”, Bioinformatics, Vol. 26, No. 3, pp. 302-309, 2010.
- R. Gur-Arie, C.J. Cohen, Y. Eitan, L. Shelef, E.M. Hallerman, and Y. Kashi, “Simple sequence repeats in Escherichia coli: Abundance, distribution, composition, and polymorphism,” Genome Research, Vol. 10, pp. 62-71, 2000.
- R. Seth, L. Sun, C.K. Ea, and Z.J. Chen, “Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3,” Cell, Vol. 122, pp. 669-682, 2005.
- O. Takeuchi and S. Akira, “MDA5/RIG-I and virus recognition,” Current Opinion in Immunology, Vol. 20, pp. 17-22, 2008.
- D. de Nardo and E. Latz, “NLRP3 inflammasomes link inflammation and metabolic disease,” Trends in Immunology, Vol. 32, pp. 373-379, 2011.
- H. Yanai, D. Savitsky, T. Tamura, and T. Taniguchi, “Regulation of the cytosolic DNA-sensing system in innate immunity: a current view,” Current Opinion in Immunology, Vol. 21, pp. 17-22, 2009.
- G.L. Horvath, J.E. Schrum, C.M. de Nardo, and E. Latz, “Intracellular sensing of microbes and danger signals by the inflammasomes,” Immunological Reviews, Vol. 243, pp. 119-135, 2011.
