Application of the Docking Protocol Optimization for Inhibitors of IGF-1R and IR and Understanding them through Artificial Intelligence and Bibliography

Author: Mustafa Kamal Pasha, Khurram Munawar, Asma Talib Qureshi
Journal: International Journal of Education and Management Engineering @ijeme
Article in issue: 4 vol.11, 2021.
Free access
The cancer cell prolonged and continues proliferation is a major cause of tumorigenesis. In general, Insulin like growth factor receptor (IGF-1R) and Insulin receptor (IR-A) protein are responsible for such cell proliferations. However, with respect to cancers, the specific over-expression of these receptors along with the elevated levels of their agonist, i.e. insulin-like growth factor 1 (IGF-1) and insulin-like growth factor 2 (IGF-2) have shown to be the integral part of cancer cell’s proliferation. The understanding of the dual targeting of (IR) and (IGF-1R) through Artificial Intelligence in tumorigenesis is now considered to be a possible aspect to achieve the desired results. In this research we signify that according to data based on artificial intelligence, the tyrosine kinase domain of these two receptors can accommodates number of small molecules inhibitors to block the ongoing signaling cascade for cell proliferation. It is indeed found to be of paramount importance to develop such candidates as clinical solutions to block the activity of tyrosine kinase domain of IR and IGF-1R. Therefore, this study aims to use artificial intelligence for understanding the key molecular interactions responsible for activation and inhibition of the proliferation signal via tyrosine kinase domain. Further, we optimized docking protocol on crystal structures of such system from protein databank. Our study revealed that H-bond donor and hydrophobic pocket play a key role in the initiation of the signal cascade for cell proliferation. The simulations ran produced an acceptable solution based on the statistical measures of Mathew’s correlation factor and delineated two H- bonds distances between 12-22. Our study also concluded that how a docking protocol can be optimized to accommodate the non-congeneric series small molecules. We successfully ran the simulation to conclude that LYS 1030, GLU 1077, MET 1079 and ASP 1083 amino acids positions play an important role in binding of small molecules to inhibit cancer cell proliferation. This research bridges the gap between in-silico and in-vitro experimentations and paves a way to reproduce the results experimentally.
Rtificial intelligence, Molecular modelling and docking, IGF-1R, IR, Dual Inhibitors, Molecular Interactions, Tyrosine Kinase Domain, Docking.
Short address: https://sciup.org/15017850
IDR: 15017850 | DOI: 10.5815/ijeme.2021.04.01
Text of the scientific article Application of the Docking Protocol Optimization for Inhibitors of IGF-1R and IR and Understanding them through Artificial Intelligence and Bibliography
Published Online August 2021 in MECS DOI: 10.5815/ijeme.2021.04.01
In the datasets collected and filtered through artificial intelligence, IGF-IR and IR fall in receptor tyrosine kinases (RTKs) and their normal physiological function is cell growth and survival. Inhibition of any of these targets results in the stoppage of cancer cell proliferation. We found that initially, studies have focused only on screening and developing small molecule inhibitors against the IGF-1R only but targeting IGF-1R alone can have undesirable consequences and may lead to implications that may strengthen malignancies [6]. Also, targeting IR-A alone would result in hyperglycemia, leading to metabolic complications [7]. Co-targeting both the receptors in order to make sure that the mitogenic signaling cascade activated by both these receptors is down-regulated so that proliferative pathways that lead to tumor formation can be stopped [3]. Since the establishment of the fact that both receptors are responsible for cancer progression and can be targeted simultaneously [4], there came studies revealing that the drug binding site affinities are different for these receptors [5]. Interestingly, it has been reported that the ATP binding site of both IR and IGF-1R have a high degree of sequence similarity and this pocket has been targeted in a number of studies [5]. We objectified to perform docking studies in the tyrosine kinase domain of these receptors to provide a realistic target approach for small molecular inhibitors. This led us to believe that there is sufficient evidence for Insulin Receptor-A (IR-A) role in transformation and maintenance of tumors [8]. Our studies reduced the limitation of the results by only using the latest crystal structure of Insulin receptors available on protein database portal. Targeting one receptor, i.e. IGF-1R, may have undesirable consequences and lead to insinuations that may strengthen malignancies through compensatory mechanisms [8]. Therefore, the increasing evidence that repressing any of the RTKs is not enough, the still functional parallel receptors will do enough to compensate the cross-talks and the system persists in tumor state. There also exists Corroboration that IGF-II binds to IR-A resulting in higher level of mitogenic signaling [9]. Upon attachment of IGF-II the kinetic domain of IR-A is structurally displaced bringing the beta domains closer for phosphorylation as explained in Fig. 1. Therefore, we targeted the simulation to co-target both the receptors in order to make sure that the mitogenic signaling cascade activated by both these receptors is down-regulated so that proliferative pathways that lead to tumor formation can be stopped [3].
Fig. 1 The Insulin Receptor with Insulin A: Attachment of insulin results in structural change which brings β domain closer and Phosphorylation takes place, B: The Apo state (or basal) where there is no phosphorylation prior to attachment of insulin [10].
Although, the insulin receptor (IR) is involved in the metabolic pathways in routine but there exist previous studies point towards its high expression in cancer. Particularly, when small molecule inhibitors are targeted against IGF-1R, it has been observed that the tumours switch towards IR as an alternative to keep up with the proliferative pathways and the mitogenic cascades. The extent of similarity is quite high and has been reported to be around 45-65% in the ligand binding domains and approximately 60-85% homology exists in the tyrosine kinase domains [11]. Co-targeting was started as early as in 2006, when Haluska et al. reported in vivo and in vitro proves of dual inhibition of both receptors with BMS-554417 [12]. Recently, it has been suggested that dual targeting of receptors could be helpful to evade from compensatory crosstalk as a survival mechanism generated in the presence of solo target i.e. IGF1R. Therefore, giving bigger therapeutic window and beneficial response. This being blocked, the unwanted proliferation and subsequent tumor survival will subside [4] . Therefore, in order to identify the best key molecular interactions responsible for activation and inhibition of the proliferation signal via tyrosin kinase domain, this study aims to collect data through artificial intelligence. The docking protocol optimization of such system has been also explained in this study through artificial intelligence.
Artificial intelligence approach is preferred in this study, owing to the increased use of computers and internet sources, for the vast volumes of data [13, 14]. Other than computer, the devices like antennas and wireless devices can also be used for the collection of data. As the data collected through these devices would have large benefits, such as wearable antennas are now being used in personal wellbeing, entertainment, military and biological sciences [15- 21].
2. Methodology 2. 1 Data Collection
A diverse dataset of 33 highly potent dual inhibitors IGF-1R and IR with known inhibitory potency (IC50) values were extracted from ChEMBL literature database [22]. Keywords used in this data base specifically for dual inhibition were related to the research domains, as shown in Fig. 2.

mpdel dockirtg study syndesis
Fig. 2 Keywords found in articles related to molecular docking.
molecular dynamic
The dataset belonged to the diverse classes of compounds, i.e., imidazopyrazine, imidazopyridine, pyrrolopyramidine, quinolines, cyanoquinolines and isoquinolinedione series [2, 23]. This IC 50 values of this data was converted into PIC 50 values for data simplification using the Equation 1. Inconsistencies in the dataset were removed by the application of data refinement steps, which are explained in Fig. 3.
PICS0 =- igicso
Fig. 3 Data refinement steps based on Artificial Intelligence (AI)
Fig. 4 Graphical Abstract: Integrated Docking and pharmacoinformatics approach for the identification of conventional and non-conventional ligand interactions.
The 3D structure of finally selected 33 potent inhibitors as provided in supporting information collected were generated through artificial intelligence from smiles (simplified molecular-input line-entry system), which is a specification in the form of line notation for a chemical structure using short ASCII string, using MOE the structure as shown in Fig. 4.
For a rational drug design and discovery, we performed structure-based drug design as explained in Fig. 2. The main objective of Structure based drug design is to see designed drug molecule binding a target (Protein or Amino acids). This further clarifies the in depth understanding of the nature of binding affinity and identifies the amino acids responsible for eliciting the pharmacological responses. The sole idea of performing a structure-based docking strategy is to conceive the electrostatic and steric hindrances to find out the highest receptor affinity. The diligent inspection of the available 3-D structure enables us to visualize and inspect the binding site, topology, affinity and protein residual interactions including the sub-pockets. Major focus has given to carefully examine important features and efficient modelling optimization for the target receptor.
-
2. 2 Molecular Docking
Besides the extraction of small molecule inhibitors, a 3D receptor structural search was also done which gave different structures that could be used in the analysis. AI based analysis framework of molecular docking is shown in Fig. 5.
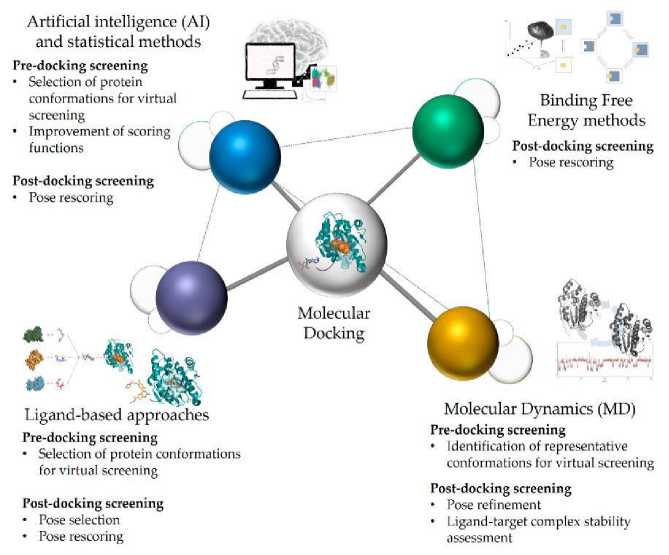
Molecular Docking
Binding Free Energy methods
Post docking screening
* Pose rescoring
Artificial intelligence (AI) and statistical methods
Pre-docking screening
* Selection of protein conformations for virtual screening
• Improvement of scoring functions
Post-docking screening
* Pose rescoring
Ligand-based approaches
Pre-docking screening
• Selection of protein conformations for virtual screening
Molecular Dynamics (MD) Pre-docking screening
* Identification of representative conformations for virtual screening
Post-docking screening
* Pose refinement
♦ Ligand-target complex stability assessment
Post-docking screening
• Pose selection
• Pose rescoring
Fig. 5 Structural representation of Molecular Docking through AI.
From this crystallographic database of receptors, the structural comparison of the tyrosine kinase domain of IR and IGF1R was done (Table 1). After matching the pocket residues and loop comparison, it was critical to choose IGF1R and IR structures for analysis based on their geometry and 3D coordinates. For this, these structures were aligned and blossom62 [24] matrix-based calculation of root mean square deviation (RMSD) was performed. Moreover, it was also considered that whether the Co-crystal structures are recently published and have higher resolution. On account of above comparison, PDB ID 5HZN of IGF1R [1] and PDB ID 4IBM [2] of IR were selected.
Table 1 Structural Database Comparison of IGF-1R and IR
|
IR |
IGF1R |
RMSD |
Chain similarity [%] |
|
4IBM [2] |
3D94 [ 25 ] |
3.535 |
77.9/76.5 |
|
4IBM [2] |
3I81 [ 26 ] |
2.575 |
77.4/ 77.9 |
|
4IBM [2] |
3WL0 [ 27 ] |
1.824 |
77.4/ 76.9 |
|
4IBM [2] |
4D2R [ 28 ] |
2.539 |
78.3/79.9 |
|
5ES1 [42] |
3I81 [ 29 ] |
2.183 |
75.7/78.9 |
|
5ES1 [42] |
3WL0 [ 30 ] |
2.036 |
75.7/77.8 |
|
5HHW [1] |
3I81 [ 31 ] |
2.388 |
79.4/76.8 |
|
5HHW [1] |
3WL0 [ 32 ] |
2.065 |
80.5/76.8 |
-
2. 3 Docking and Pose Analysis through Artificial Intelligence
Dock application from MOE was used to produce optimal fit configurations of the inhibitors and macromolecular target (IGF-1R & IR). For each inhibitor from the database, a set of conformations were generated and scored in an effort to determine favourable binding mode. Prior to docking the database, a site was required to be found for ligands to bind. For this, A grid representation of the molecular volume by Hendlich et al. [33] to locate the binding site for ligand was performed. A built-in application of the MOE known as “Site Finder” based on Alpha Shapes was used to determine the binding pocket automatically[33].
The first choice for docking was to perform runs consecutively with each member of ensemble by using rigidreceptor docking. The in such a way that it could be used for different stages which could be integrated into the framework, the framework is shown in Fig. 6. The dock algorithm [34]automatically generated 3D conformations which seemed to be optimal fit into the pocket with the help of ‘Placement method’ for ligand placement
Fig. 6 Framework for the optimization docking protocol through AI
Following placement methods were used in the optimization of protocol
-
• Alpha Triangle
-
• Triangle Matcher
-
• Alpha PMI
After the placement of the ligand into the pocket it was then scored having stress on favourable ionic, hydrophobic and H-bond contacts. The variability of the scoring functions as determined by Corbeil et al. [35] was considered. The lower the score, the better the pose was considered. Following scoring functions were used
-
• London dG Scoring
-
• Affinity dG Scoring
-
• Alpha HB Scoring
-
2. 4 Consensus Scoring
Upon several combinations of placement methods and scoring functions for docking; Alpha PMI and Alpha HB was considered final for further dockings and analyzation. Once Docking runs were performed the generated databases of optimal conformations for bioactive interactions between molecules and receptors were further analysed via consensus scoring. It was then used for GRIND, QSAR, Pharmacophore and virtual Screening.
After the dock application run for database (Table 1) a large conformational space was produced via particular scoring function and placement method. The whole of the information was transferred into excel spread for analysis, scoring methodology is presented in Fig. 7.
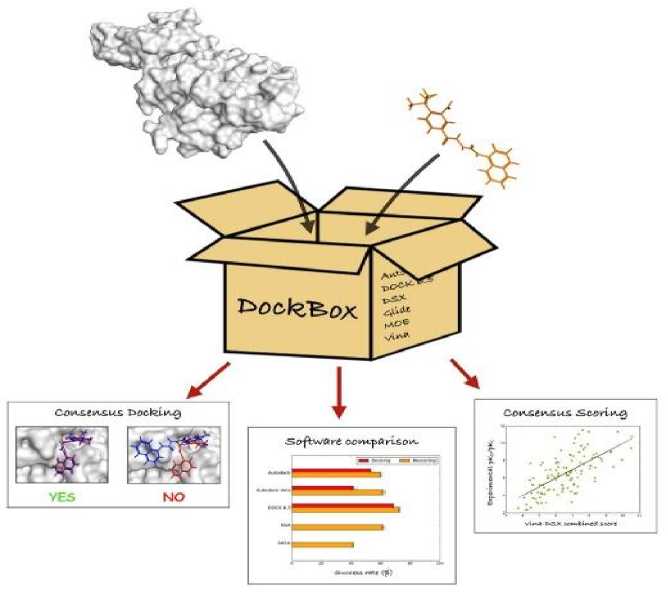
Fig. 7 AI based Methodology for Scoring
It has been reported by Charifson et al. [36] that 3-4 scoring functions are sufficient for deducing good results. The poses generated were sorted according to their relative scores (rank-by-number). Different scoring functions were used again for ranking the same conformational space. After all the scoring functions were utilized for the same space, they were ranked according to their scores assigned later (rank-by-rank), as shown in Fig. 8. Finally, the average was taken for each entry and then the ranking was performed again on the basis of average voting for top 10% to produce final rank upon which decision could be made (rank-by-vote).
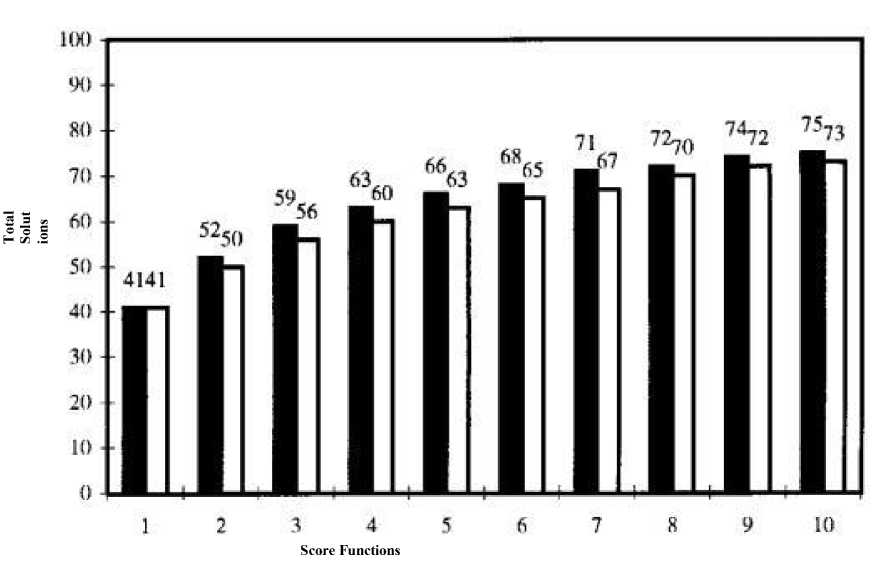
Fig. 8 Relationship between scoring functions used in consensus scoring (X-axis) and the total solution (Y-axis). Solid bars were for rank-by-number and hollow bars were for rank-by-rank strategy.
For Vander Wal (Vdw) and Hydrogen boding (H-bonding) interactions following 2 equations were utilized.
к-r = ∑ , ∈ • ∑ , ∈ 1 [( dtj ) -( dtj )] (2)
K^D^ = ∑ A ∑ D [(^) -2( T^ )]×cos2( e - e0) (3)
LVu^-D/ VdA_D; J
Where, dA-D represents the distance between a hydrogen bond acceptor (HA) and a hydrogen bond donor (HD)
θ is the bond angle between HD and HA r0 and θ0 denote the optimal distance and angle for a specific pair of HA and HD which, are dependent on the types of donor atom and acceptor atom.
Following assumptions were taken into account before the rank analysis.
• All the compounds were docked perfectly
• For convenience, it was ruled that each scoring function had accuracy at the same level for the database.
• Each scoring function was independent to each other.
3. Result and Discussion
3.1 Docking Pose Selection
Docking was performed on a energy minimised dataset of dual inhibitors. The 3D conformations of inhibitors were minimized by calculating atomic coordinates that are local minima of the energy function using molecular mechnics and forcefield MMFF94 based algorithms [37]. The pdb structure 5HZN (IGF1R) used as atarget was also minimized by suing the same force field in a similar fashion. Upon several combinations of placement methods and scoring functions for docking; Alpha PMI and Alpha HB was considered final for further dockings and analyzation. The binding site recognition causes the identification and characterisation of the small molecular interactions in the domain with a fixed binding affinity. This gives us further possible binding poses and also active sites.
Table 2 Placement methods and scoring functions combinations used in docking
|
S. No |
Placement Method |
Scoring Fn. |
E_Score |
RMSD (Å) |
Rank |
Residues involved |
|
1. . |
α PMI |
Affinity dG |
-5.0194 |
2.0704 |
1 |
Lys 1030 |
|
2. |
α PMI |
α HB (Chosen) |
-116.528 |
2.3095 |
6 |
Lys 1030, Phe 1151, Glu 1077 |
|
3. |
α PMI |
London dG |
-15.0063 |
2.3095 |
1 |
Lys 1030, Phe 1151, Glu 1077 |
|
4. |
α Triangle |
Affinity dG |
-3.7146 |
2.4203 |
1 |
Lys 1030 |
|
5. |
α Triangle |
α HB |
-133.5868 |
1.4658 |
3 |
Lys 1030^^^^H |
|
6. |
α Triangle |
London dG |
-16.1993 |
2.0046 |
56 |
Lys 1030, Phe 1151, Gly 1152 |
|
7. |
Triangular matcher |
Affinity dG |
-3.1628 |
2.1076 |
7 |
Lys 1030 |
|
8. |
Triangular matcher |
α HB |
-113.7775 |
2.6610 |
5 |
Lys 1030, Glu 1077 |
|
9. |
Triangular matcher |
London dG |
-112.6802, -67.2396 |
2.2002, 2.4583 |
1, 41 |
Lys 1030, Arg 1136--- Lys 1030 |
Dock application[29] from MOE was used to produce optimal fit configurations of co-crystallized inhibitor (NVP-AEW541) and macromolecular target (5HZN). A set of conformations were generated and scored in an effort to determine favourable binding mode. The placement of the ligand (small molecule) finds the possible orientation and interactions. Three different placement methods and scoring functions were utilized in an effort to produce optimal fit that best matches with the co-crystallized conformation and also the Interactions. The co-crystal information was matched against the final pose obtained via docking (Fig. 9) of the similar compound. In addition to Lys 1030, new interactions i.e. Phe 1151, Glu 1077forming hydrophobic interactions were found. Almost 900 conformations with different placement methods i.e. Alpha Triangle, Triangle Matcher, Alpha PMI, were generated which were found to be optimally fit into the pocket of the receptor. Later, these conformations were scored via scoring function algorithm based on favourable ionic, hydrophobic and H-bond contacts and the variability was considered [30]. Poses having lower scores were considered for further analysis.
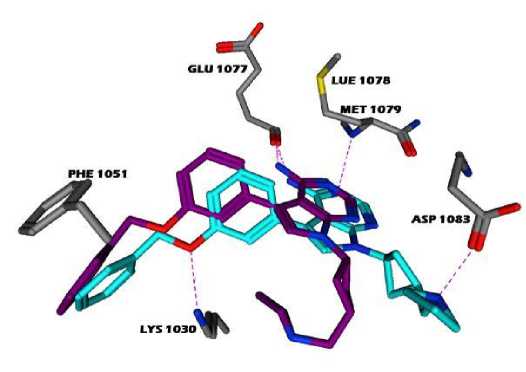
Fig. 9 Co-crystal (Blue) and docked (Purple) conformation interaction pattern.
This combination was used to perform docking runs for the database of dual inhibitors. The pose generation threshold for each inhibitor was set to be 100. The generated databases of optimal conformations for bioactive interactions between molecules and receptors were further analysed via consensus scoring. The product of docking runs was rescored in order to extract the information of the best bioactive conformation. Two step binding conformation requirements included: (i) examination of a larger conformational space with various potential binding modes; (ii) Predicting the energy interaction of the binding conformation [24] .
3.2 Final Pose Selection
4. Conclusion
An excel spread analysis was performed on the solutions produced by the docking optimization. It was already published approach to use 3-4 scoring functions. Ranking was performed according to the relative scores via rank-by-number approach, as shown in Fig. 10. Later, rank-by-rank approach is used for the second rescoring function [35]. Finally, the ranking on top 10% was performed via rank-by-vote for each entry based on the consensus produced by the average taken against each entry.
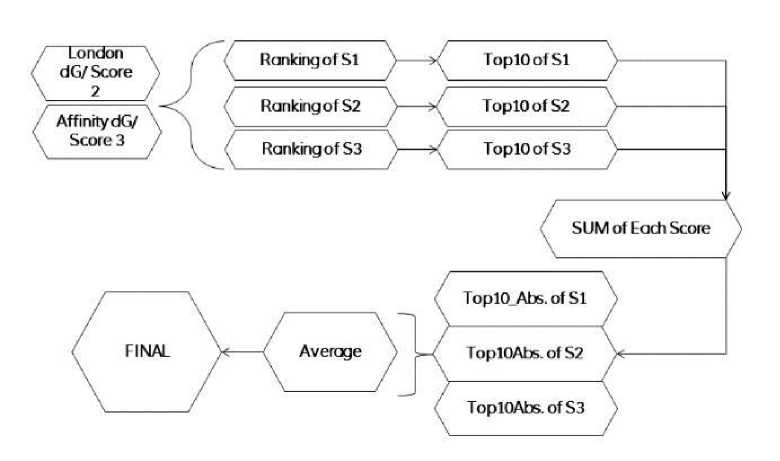
Fig. 10 Consensus scheme used to produce best solutions. S1*= scoring scheme named α-HB used previously while redocking the co-crystal ligand. S2*= London dG. S3*= Affinity dG.
A total of 1187 poses were generated for the curated database by assigning threshold of 100 poses per ligand. They were then ranked according to their least energy. Top 10 poses were given “1” and “0” was assigned to rest of solutions. A total sum of top 10 solutions was taken and poses having rank by vote as 3/3 were considered most suitable. A consensus was built upon average of the poses having maximum votes and least rank. Hence, this is found as the best optimization techniques for the doping procedures.
Artificial Intelligence can help a lot in understanding the Docking Protocol Optimization for Inhibitors of IGF-1R and IR [38-39]. The best docking protocols will result in greater and better results of biological experiments. The use of innovative technology such as machine learning, artificial intelligence will help apply these protocols in biological sciences efficiently [40-42]. Evaluating the poses and ranking them based on their score gave us the optimum number of selected poses to be further analyzed.
The docking results concurrently delineated two H-bond donors, two H-bond acceptor, overall Topology and vdw_vol (Wander Val force) as important descriptors for dual inhibition of IGF-IR and IR. Moreover, it showed two H-bond donors at the distance of 16.8-17.2 Å and two H- bond acceptors at the distance of 12.8- 13.2 Å respectively to be important for the domain selectivity and affinity. It also predicted steric bulk along with H- bond donor at 18-22 Å. Furthermore, our pharmacophore model delineated the distances between the descriptors like hydrophobic region, H-bond acceptors and donor region to be of crucial importance in terms of binding dual inhibitor. Mathew’s correlation factor of our pharmacophore model was 0.87 based on TP hit rate. Since the dataset belonged of IGF-IR and IR inhibitors belonged to the non-congeneric series which belonged to diverse set of classes, it proved that the optimization of the docking protocol can lead us to the discovery of novel molecules. However, presently used algorithms and estimations do not provide a final decree in terms of intermolecular interactions with absolute accuracy. The docking methodology has an ability to screen large number of conformational spaces at low cost including high throughput screening. Even then, further artificial intelligence-based studies may however can prove vital for the drug discovery and optimisation of small molecules. This would not only require iterative virtual screening methods but also an improved translational methodology scheme is needed for future studies. Recent advances in molecular dynamics (MD) simulations can further investigate the poses and interactions to find the suitability of the researched pose in this study.
References Application of the Docking Protocol Optimization for Inhibitors of IGF-1R and IR and Understanding them through Artificial Intelligence and Bibliography
- Stauffer, F., et al., Identification of a 5-[3-phenyl-(2-cyclic-ether)-methylether]-4-aminopyrrolo [2, 3-d] pyrimidine series of IGF-1R inhibitors. Bioorganic & medicinal chemistry letters, 2016. 26(8): p. 2065-2067.
- Anastassiadis, T., et al., A highly selective dual insulin receptor (IR)/insulin-like growth factor 1 receptor (IGF-1R) inhibitor derived from an extracellular signal-regulated kinase (ERK) inhibitor. Journal of Biological Chemistry, 2013. 288(39): p. 28068-28077.
- Huang, F., et al., IRS2 copy number gain, KRAS and BRAF mutation status as predictive biomarkers for response to the IGF-1R/IR inhibitor BMS-754807 in colorectal cancer cell lines. Molecular cancer therapeutics, 2015. 14(2): p. 620-630.
- Jin, M., et al., Small-molecule ATP-competitive dual IGF-1R and insulin receptor inhibitors: structural insights, chemical diversity and molecular evolution. Future medicinal chemistry, 2012. 4(3): p. 315-328.
- Vincent, E.E., et al., Targeting non-small cell lung cancer cells by dual inhibition of the insulin receptor and the insulin-like growth factor-1 receptor. PLoS One, 2013. 8(6): p. e66963.
- Yamaguchi, Y., et al., Ligand-binding properties of the two isoforms of the human insulin receptor. Endocrinology, 1993. 132(3): p. 1132-1138.
- Munshi, S., et al., Structure of apo, unactivated insulin-like growth factor-1 receptor kinase at 1.5 Å resolution. Acta Crystallographica Section D: Biological Crystallography, 2003. 59(10): p. 1725-1730.
- Buck, E., et al., Compensatory insulin receptor (IR) activation on inhibition of insulin-like growth factor-1 receptor (IGF-1R): rationale for cotargeting IGF-1R and IR in cancer. Molecular cancer therapeutics, 2010. 9(10): p. 2652-2664.
- Denley, A., et al., The insulin receptor isoform exon 11-(IR-A) in cancer and other diseases: a review. Hormone and Metabolic Research, 2003. 35(11/12): p. 778-785.
- De Meyts, P. and J. Whittaker, Structural biology of insulin and IGF1 receptors: implications for drug design. Nature Reviews Drug Discovery, 2002. 1(10): p. 769-783.
- Torres, A.M., et al., Solution structure of human insulin-like growthfactor II. Relationship to receptor and binding protein interactions. Journal of molecular biology, 1995. 248(2): p. 385-401.
- Haluska, P., et al., In vitro and in vivo antitumor effects of the dual insulin-like growth factor-I/insulin receptor inhibitor, BMS-554417. Cancer Research, 2006. 66(1): p. 362-371.
- Munawar HS, Awan AA, Khalid U, Munawar S, Maqsood A. Revolutionizing Telemedicine by Instilling H. 265. International Journal of Image, Graphics & Signal Processing(IJIGSP). 2017 May 1;9(5).
- Munawar, H. S., Qayyum, S., Ullah, F., & Sepasgozar, SBig Data and Its Applications in Smart Real Estate and the Disaster Management Life Cycle: A Systematic Analysis. Big Data and Cognitive Computing; 2020, 4(2).
- Munawar, H. S., Zhang, J., Li, H., Mo, D., & Chang, L. Mining multispectral aerial images for automatic detection of strategic bridge locations for disaster relief missions. In Pacific-Asia Conference on Knowledge Discovery and Data Mining (pp. 189-200). Springer, Cham. . (2019, April)
- Munawar HS, Hammad A, Ullah F, Ali TH. After the flood: A novel application of image processing and machine learning for post-flood disaster management. InProceedings of the 2nd International Conference on Sustainable Development in Civil Engineering (ICSDC 2019), Jamshoro Pakistan 2019 Dec (pp. 5-7).
- Munawar, H. S. "International Journal of Wireless and Microwave Technologies (IJWMT)."
- Munawar, H. S. "An Overview of Reconfigurable Antennas for Wireless Body Area Networks and Possible Future Prospects."
- Munawar, H. S., Khalid, U., Jilani, R., & Maqsood, A. (2017). Version Management by Time Based Approach in Modern Era. International Journal of Education and Management Engineering(IJEME), 4, 13-20.
- Munawar, H. S. "Reconfigurable Origami Antennas: A Review of the Existing Technology and its Future Prospects."
- Munawar, H. S., & Maqsood, A. Isotropic Surround Suppression based Linear Target Detection using Hough Transform.
- Gaulton, A., et al., ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic acids research, 2011. 40(D1): p. D1100-D1107.
- Jin, M., et al., Discovery of an orally efficacious imidazo [5, 1-f][1, 2, 4] triazine dual inhibitor of IGF-1R and IR. ACS medicinal chemistry letters, 2010. 1(9): p. 510-515.
- Henikoff, S. and J.G. Henikoff, Amino acid substitution matrices from protein blocks. Proceedings of the National Academy of Sciences, 1992. 89(22): p. 10915-10919.
- Wu, J., et al., Small‐molecule inhibition and activation‐loop trans‐phosphorylation of the IGF1 receptor. The EMBO journal, 2008. 27(14): p. 1985-1994.
- Wittman, M.D., et al., Discovery of a 2, 4-disubstituted pyrrolo [1, 2-f][1, 2, 4] triazine inhibitor (BMS-754807) of insulin-like growth factor receptor (IGF-1R) kinase in clinical development. Journal of medicinal chemistry, 2009. 52(23): p. 7360-7363.
- Heinrich, T., et al., Allosteric IGF-1R inhibitors. ACS medicinal chemistry letters, 2010. 1(5): p. 199-203.
- Kettle, J.G., et al., Discovery and Optimization of a Novel Series of Dyrk1B Kinase Inhibitors To Explore a MEK Resistance Hypothesis. Journal of medicinal chemistry, 2015. 58(6): p. 2834-2844.
- Sanderson, M.P., et al., BI 885578, a Novel IGF1R/INSR Tyrosine Kinase Inhibitor with Pharmacokinetic Properties That Dissociate Antitumor Efficacy and Perturbation of Glucose Homeostasis. Molecular cancer therapeutics, 2015. 14(12): p. 2762-2772.
- Hendlich, M., F. Rippmann, and G. Barnickel, LIGSITE: automatic and efficient detection of potential small molecule-binding sites in proteins. Journal of Molecular Graphics and Modelling, 1997. 15(6): p. 359-363.
- Labute, P. and M. Santavy, Locating binding sites in protein structures. Journal of Chemical Computing Group, 2007.
- Knegtel, R.M., I.D. Kuntz, and C. Oshiro, Molecular docking to ensembles of protein structures. Journal of molecular biology, 1997. 266(2): p. 424-440.
- Österberg, F., et al., Automated docking to multiple target structures: incorporation of protein mobility and structural water heterogeneity in AutoDock. Proteins: Structure, Function, and Bioinformatics, 2002. 46(1): p. 34-40.
- Edelsbrunner, H., Weighted Alpha Shapes, 1992. Department of Computer Science, University of Illinois at Urbana-Champaign, Urbana, Illinois, 1992. 61810.
- Corbeil, C.R., C.I. Williams, and P. Labute, Variability in docking success rates due to dataset preparation. Journal of computer-aided molecular design, 2012. 26(6): p. 775-786.
- Charifson, P.S., et al., Consensus scoring: A method for obtaining improved hit rates from docking databases of three-dimensional structures into proteins. Journal of medicinal chemistry, 1999. 42(25): p. 5100-5109.
- Gill, P.E., W. Murray, and M.H. Wright, Practical optimization. 1981.
- Munawar, H. S., Khalid, U., & Maqsood, A. Fire detection through Image Processing; A brief overview.
- Munawar, H. S., Khalid, U., & Maqsood, A. Modern day detection of Mines; Using the Vehicle Based Detection Robot.
- Munawar HS, Awan AA, Maqsood A, Khalid U. REINVENTING RADIOLOGY IN MODERN ERA.
- Munawar, H. S. (2020). Flood Disaster Management: Risks, Technologies, and Future Directions. Machine Vision Inspection Systems: Image Processing, Concepts, Methodologies and Applications, 1, 115-146.
- Munawar, H. S. (2020). Image and Video Processing for Defect Detection in Key Infrastructure. Machine Vision Inspection Systems: Image Processing, Concepts, Methodologies and Applications, 1, 159-177.
