Applied an Efficient Site-directed Mutagenesis Method into Escherichia coli

Author: Muqing Qiu
Journal: International Journal of Information Technology and Computer Science(IJITCS) @ijitcs
Article in issue: 2 Vol. 3, 2011.
Free access
A new technique for conducting site-directed mutagenesis was developed. This method allows the color selection of mutants through the simultaneous activation or deactivation of the α-peptide of ß-galactosidase. The method can efficiently create mutations at multiple sites simultaneously and can be used to perform multiple rounds of mutation on the same construct. In this paper, in order to develop an efficient site-directed mutagenesis method in vivo, the tests were tested by the following methods. The methods that the fragment knock-out ompR gene was constructed through overlapping PCR, digested by Notand SalⅠⅠ, ligated to plasmid pKOV were applied. The recombination plasmid was transformed into Escherichia coli WMC-001 strain, integrated into the genomic DNA through two step homologous recombination. The Escherichia coli WMC-001/ompR- mutant was obtained due to gene replacement. The fragment of the mutant ompR gene was amplified through overlapping PCR, cloned into pKOV vector. The recombinant plasmid was introduced into Escherichia coli WMC-001/ompR- mutant. The Escherichia coli WMC-001/ompR mutant was also obtained due to gene replacement. Results: The site-directed mutagenesis has been successfully constructed in the ompR gene by sequencing. Conclusion: The method is effective for construction of gene site-directed mutagenesis in vivo.
Overlapping PCR, gene replacement, ompR gene, site-directed mutagenesis
Short address: https://sciup.org/15011613
IDR: 15011613
Text of the scientific article Applied an Efficient Site-directed Mutagenesis Method into Escherichia coli
Published Online March 2011 in MECS
oligonucleotide[4], reversion of a selectable marker, such as the ampicillin-resistance (ampr) gene [5], deletion of unique restriction sites to protect from digestion [6-7], reversion of an RNA binding site mutationto recover DNA replication in normal bacterial strains [8], and reduction of the background by treatment of newly synthesized DNA with the restriction enzyme Dpn I which recognizes and cleaves methylated DNA [9]. Methods using selectable markers have the advantage that they do not require additional treatments. By applying two selectable markers, such methods can also be used to perform multiple rounds of mutagenesis without recloning the target sequence [10]. However, as this method needs two selectable markers, the vector size is substantially increased, the simultaneous mutation efficiency is decreased and two separate selection steps are required to select the two markers. The use of Dpn I, although quite efficient in PCR-related site-directed mutagenesis [11], has variable results on digestion of methylated and hemimethylated DNA [12].
With the development of thermo-stable polymerases and PCR, many efforts have been made to perform and improve site directed mutagenesis using PCR methods [13-17]. Although these methods provide a quick way of obtaining a mutant DNA fragment, it is difficult to perform simultaneous multiple site mutations, and the approach often introduces additional, unwanted mutations.
One problem that is encountered with all mutagenesis methods is the need to verify that any mutant phenotype obtained is due exclusively to the desired genetic change introduced by the mutagenesis procedure. This can be done by sequencing through the entire portion of DNA that has been through the mutagenic procedure to eliminate the possibility that additional, undesired changes might be responsible for the mutant phenotype. However, this is impractical in the case of large plasmids and it is usual to sub-clone a portion of the sequence prior to mutagenesis in order to reduce the amount of subsequent sequence analysis that needs to be undertaken. An alternative approach to correlating mutant phenotype with genetic change involves the deliberate creation of revertants by back-mutation and the demonstration that these have the properties of the original sequence. This approach requires that there be an efficient way of conducting at least two rounds of mutagenesis.
Studies of UV mutagenesis have revealed that the majority of base substitution events are G:C to A:T transitions at cytosine-containing photoproducts [18]. For CPDs, it has been suggested that rapid deamination of the cytosine to uracil could allow incorporation of adenine, giving rise to the transition [19–21]. However, mutations also occur at TT sequences, giving a very different mutational pattern. Studies using TT photoproducts incorporated at specific sites have revealed a number of non-random substitution patterns. For example, Banerjee et al. [22] showed that of the base changes produced by a cis-syn TT cyclobutane dimer, 79% were 5-TT to 5-TA transversions while 17% were 5-TT to 5-TC transitions. Hence, 96% of all the mutations occurred at the 3-base of the dimer. In contrast, the trans-syn form of the TT CPD produced 24% 5-TT to 5-AT transversions and 6% 5-TT to 5-CT transitions; 69% of the mutations were double base changes, frame shifts or deletions. Thus, of the single base change mutations produced by the trans-syn CPD, 95% occurred at the 5-base of the dimer [23]. It is important to note that about 90% of the CPDs have the cis-syn conformation, while only about 10% are in the trans-syn form. In comparison, the photoproduct was roughly 10 times more mutagenic than the cis-syn CPD and it produced a very different mutational spectrum; 94% of the mutations were 5-TT to 5-TC transitions [24]. Therefore, a much greater diversity of changes are possible when considering photoproducts involving TT sequences, and the changes that occur at any particular site in the genome may reflect the contribution of all types of DNA damage.
The genome of Cowpea mosaic virus (CPMV), a plant virus, consists of two messenger-sense RNA molecules termedRNA-1 and RNA-2. RNA-1 consists of 5889 nucleotides plus a variable length of poly (A) tail and encodes the 200K polyprotein, which is processed at specific sites to give the proteins required for viral genome replication. RNA-2 consists of 3481 nucleotides and encodes the 105/95K polypeptides, which are processed to produce the viral movement protein and large and small coat proteins. Infectious cDNA clones of bothRNAs are available and it is possible to initiate an infection efficiently by agroinoculation. To increase the efficiency with which mutants of the virus can be produced and characterized, a new and simple protocol has been developed, which can be used for multiple rounds of mutagenesis using double-stranded plasmid DNA containing insertions of at least 6.4 kbp nucleotides. The method uses complementation for color selection as a screenable marker, high temperature denaturation for template preparation, T4 or T7 DNA polymerase for high fidelity DNA extension, and DpnI treatment and a mismatch repair-minus bacterial strain to reduce background. The method has been applied to full-length CPMV-based cDNA clones and the results have confirmed that it is an efficient way of introducing multiple mutations simultaneously and carrying out multiple rounds of sequential mutagenesis including the production of revertants. The method should be generally suitable for the manipulation of large DNA fragments including whole viral genomes and will enable multiple rounds of mutagenesis to be performed without the need for sub-cloning.
An additional, UV-induced photoproduct, thymidylyl-(3 to 5)-deoxyadenosine, termed theTA* photoproduct, results from the cycloaddition of the 5,6 double bonds of thymine and adenine [25-26]. Despite the fact that the quantum yield of this product was estimated to be 10–100-fold less than CPDs or [6-4] products [27], this largely unstudied minor photoproduct may have important mutagenic properties [28]. Specifically, Zhao and Taylor [28] illustrated that 82% of the plaques recovered following transfection of an SOS-induced E. coli host with a TA*-containing phage genome resulted in a targeted mutation and of these, 67% were a 5-TA to 5-TT change.
In the research of gene function, it need to change the protein through altering its nucleotide or several amino acid sequence of the genes by site-directed gene mutagenesis, so that we can observe the series of molecular or cellular structure and function changes because of this change such as in the E. coli cytoplasm, the change of the response regulator protein OmpR protein can regulate the expression of bacterial outer membrane proteins, affecting cell membrane permeability to exogenous substances, but its mechanism needs further study[29-30]. Foreign studies have shown that the codon CGC of ompR genetic coding sequence of 43th bit mutated into TGC, and made arginine becoming cysteine (R15C), promoting the expression of outer membrane protein F, C and increasing the permeability of bacteria for foreign materials [31-33]. The common technology is the site-directed mutagenesis on the function of genes and proteins, which is mostly based on PCR technology or mutating a certain point of plasmid fragment [34]. In order to direct ompR gene to mutate in the E. coli genomes, without other marks of any exogenous bacterial genome, we designed a kind of effective method of site-directed mutagenesis of E. coli genome ompR gene in vivo. Its principles and programs are as follow: the ompR gene was knockouted through using the shuttle characteristics of pKOV plasmid and the principle of homologous recombination, and then E. coli ompR mutations were obtained through restructuring the mutations ompR gene (R15C) into the Escherichia coli WMC-001/ompR-mutant. This method can effectively and accurately carry out site-directed mutations in vivo.
-
II. M ATERIALS AND METHODS
-
A. Materials
Strains and plasmids: E.coli WMC-001 is constructed and saved by the lab. pKOV plasmid is provided by the Louisiana State University, with the size of 8673bp, chloramphenicol resistance, temperature sensitivity and contain SacB gene for sugar-sensitive. E.coli WMC-001 was cultured in LB medium chloramphenicol(20ug/ml) at 30o C . When the culture temperature is 42 X , the recombination strain by plasmid and chromosomal can be proliferation, and host bacteria containing free plasmid are not proliferation. The recombination plasmid was transformed into Escherichia coli WMC-001 strain, integrated into the genomic DNA through two step homologous recombination in the LB
medium containing sucrose (5%w/v) chloramphenicol at 30 X . The clones with sucrose resistance and chloramphenicol-sensitive were screenedfor further analysis.
Reagents: Restriction enzyme Sal I , Not I , Pyrobest DNA polymerase, T4 DNA ligase, plasmid extraction kit a small amount, PCR product purification kit, gel extraction kit were purchased from Takara Biotechnology Co., Ltd.. Restriction enzymes Af Ш enzyme is the products of NEB.
B. Methods
Pinciple: the principle is homologous recombination using the plasmid pKOV of temperature-sensitive, shown in Figure 1.
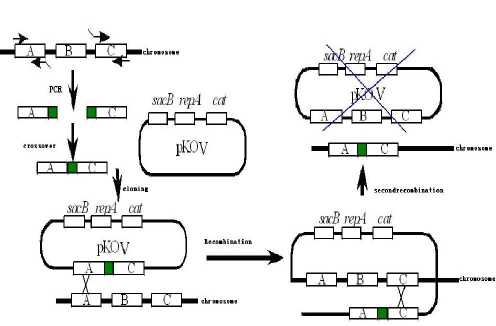
Fig.1 Schematic diagram of homologous recombination using the plasmid pKOV
PCR of primer design: according to and biological, and combing with software, the designed primers are the following table1.
Table 1 PCR of primer design
primer Base sequence
remarks
Ni
5* cacgcaataaccttcacactccaaatttataaccattgtttgtactcccaaaggttc 3'
Shadow bases
complementary tail
Crossed for the Not
No
5' aaggaaaaaagcggccgcccacgcgccaaaaaagactttg 3'
restriction site
Shadow bases
Ci
5' gttataaatttggagtgtgaaggttattgcgtgctacgtctttgtaccggacgg 3'
complementary tail
Crossed for the Sa?
Co
5' cgcacgcatgtcgacaaatcgccctgatgaatttcggt 3'
restriction site
Nm
5' ttccagcagcgcacgcaggca(g)catgtcgtcatcgacca 3'
The original sequence
OmpR gene was knockouted: The N-terminal part of NR of ompR gene containing Not I restriction site was amplified by using E.coli WMC-001 genomic DNA as template and the upstream and downstream primers Ni, No respectively, and the C-terminal part of CR of the ompR gene containing Sal I restriction site was amplified through using the upstream and downstream primers Cm, Co respectively; then the product of ompR-gene fragment was amplified through using NR, CR as templates and No, Co for primers, and PCR products were recycled and purified. PCR conditions were: 94 X 1 min; 88 X 4 min; 94 X 10 sec, 66 X 3 min, 25 cycles; 72 X 10 min, 4 X saved. The ompR-gene fragment and plasmid pKOV were digested with restriction enzymes Not I, Sal I for 4h, and the digestion products were confirmed by using 1kb DNA marker as a control, recycled and purified. The proper ligation products were linked into E.coli WMC-001 competent cells by using T4 ligase overnight (16 X ), and the bacteria was cultured in the LB plate containing chloramphenicol (20p.g/ml) at 30 X . After 48h, the colonies were picked and expanded culture, then plasmids pKOV-ompR- were extracted, and recombinant cloning strains were identified by digested. The Escherichia coli WMC-001/ompR- mutant was successfully obtained by the plates containing chloramphenicol (20μg/ml) screening at 42 X , the plates containing 5% sucrose / chloramphenicol screening at 30 X and chloramphenicol negative screening. The ompR- mutant strain genomic were identified by PCR[35].
OmpR-missing Preparation of competent bacteria [36]: The N-terminal part of NR of the ompR gene containing Not I restriction site was amplified by using E.coli WMC-001 genomic DNA as template and the upstream and downstream primers Nm, No respectively, and the C-terminal part of CR of the ompR gene containing Sal I restriction site was amplified through using the upstream and downstream primers Cm, Co respectively; then ompR -gene fragment was amplified through using NR, CR as templates and No, Co for primers, and the condition of PCR as before. The ompR mutation fragments were confirmed by Afl Ш digestion and sequencing. The confirmed fragments of ompR gene mutation were digested with restriction enzymes Not I , Sal I , linked to the pKOV which was digested with the same restriction enzymes by using T4 ligase enzyme, and transformed into competent cells of ompR -deletion strain and screened ompR mutation strain as before. The ompR mutation genomic was identified by PCR and sequencing.
-
III. R ESULTS
-
A. OmpR gene was successfully knockouted
The product of the knocked out ompR gene, about 1067bp, was successfully amplified through overlapping PCR, and the expected fragments of the recombination plasmid pKOV-ompR- digested by Not I and Sal I were about 1067bp and 5638bp respectively. Lane 1 was the positive recombination plasmid pKOV-ompR digested by
Cm
5' tggtcgatgacgacatgt(c)gcctgcgtgcgctgctggaa 31
in brackets
Bracket base before the
mutation
Not I and Sal I ; Lane 2 was the product, 1067bp, of ompR- gene; Lane 3 was the undigested recombination plasmid pKOV-ompR-( Fig.2).
The 1067bp fragment was amplified through the genome of the mutant ompR bacteria as a template and No and Co as a template (Fig.3 Lane 3); the about 1716bp fragment was amplified through the genome of E.coli WMC-001 as a template and No and Co as a template (Fig.3 Lane 2); Lane 1 was the PCR negative control; so we successfully knocked out the ompR gene as we expected.
bp
M 1 2 3
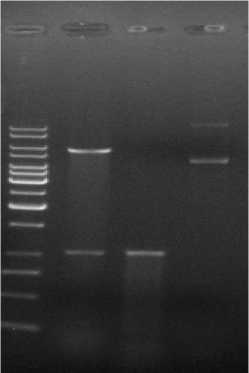
Fig.2 Identification of the recombinant plasmid pKOV-ompR- digested by Sal I and Not I
Lane 4) and the fragments,1113bp and 556bp, were digested by АА Ш (Fig.4 Lane 2 and 3). The site-directed mutagenesis has been successfully constructed in the ompR gene by sequencing.
bp
M 1 2 3
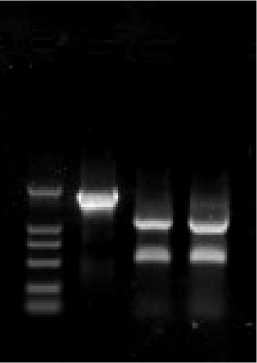
Fig.4 Identification of the fragment of the mutant ompR gene
C. The successful obtaining of the mutant ompR gene
The 11716bp fragment was amplified through the genome of the mutant ompR bacteria as a template and No and Co as a template. The site-directed mutagenesis successfully constructed in the ompR gene has been confirmed by sequencing of cloned to T vector.
bp
M 1 2 3
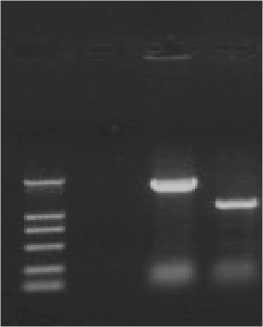
Fig.3 PCR analysis of E.coli WMC-001 and mutant E.coli WMC-001/ ompR
-
B. Identification of the fragment of the mutant ompR gene
The product of the fragment of the mutant ompR gene amplified through overlapping PCR located in about 1716bp through 1% agarose gel electrophoresis (Fig.3
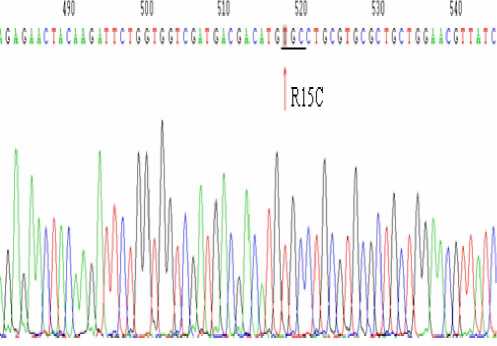
Fig.5 The sequence map of the mutant ompR gene
Number equations consecutively. Equation numbers, within parentheses, are to position flush right, as in (1), using a right tab stop. To make your equations more compact, you may use the solidus ( / ), the exp function, or appropriate exponents. Italicize Roman symbols for quantities and variables, but not Greek symbols. Use a long dash rather than a hyphen for a minus sign. Punctuate equations with commas or periods when they are part of a sentence.
-
IV. D ISSCUSSION
The present study has produced an efficient and rapid site directed mutagenesis method by using complementation for color selection as a screen able marker, together with Dpn I digestion to reduce the unmutated background, a mismatch repair free bacterial strain to maintain mutated strands and short incubation and retransformation to achieve homogeneity. This protocol allows the introduction and selection of desired multiple mutants in as little as 24 h.
Nature has produced an impressive assortment of hydrolytic enzymes belonging to nearly 85 families of glycosidases to cope with the large number of chemically and structurally different glycosides (for more information, visit the Carbohydrate Active Enzymes web site server at .
Since the time when the crystal structure of lysozyme was resolved (Phillips, 1966), the catalytic mechanisms of glycosidic enzymes have been studied intensively. The catalytic mechanism of chitinase was first suggested to be similar to that of hen’s egg white lysozyme, which utilizes a carbonium ion intermediate . With more structural data available regarding different chitinases, however, other hypotheses concerning catalytic mechanisms were proposed including a singledisplacement “retaining” reaction with an oxocarbenium ion transition state (TS), a “retaining” doubledisplacement reaction with an oxazoline TS, or an “inverting” double-displacement reaction involving both an oxocarbenium ion and abound water in the TS . Bombyx mori chitinase, which is closely related to M. sexta chitinase in sequence and other properties, was found to utilize a “retaining” mechanism, producing products that retain the β-anomeric configuration. All of the enzymes of this family examined so far are inhibited by allosamidin, an aglycone allosamizoline analog of oxazoline, which is the transition state intermediate proposed for family 18 chitinases. Additional evidence supporting such a mechanism was obtained from the crystal structures of two group 18 chitinases and four other related proteins. The crystal structure of a complex between allosamidin and hevamine, also a family 18 chitinase, revealed that loops at the carboxyl ends of the two beta-sheets containing the two conserved regions form part of the substrate binding cleft. In fact, the conserved glutamate is very near to sub-sites 1 and +1 in the binding cleft. M. sexta chitinase contains the two conserved signature sequences of family 18 chitinases and is inhibited by allosamidin, suggesting that it very probably utilizes a “retaining” double displacement reaction with an oxazoline TS. Results obtained from homology modeling of M. sexta chitinase also are consistent with such a reaction mechanism. Recently, a new modified “substrate assisted” catalysis mechanism for chitinolytic enzymes such as insect chitinase was proposed where a water molecule that is hydrogen bonded to the NH of the acetamido group also participates in the hydrolysis. Previously, the roles of glutamic and aspartic acids in conserved region II of five other family 18 chitinases were investigated, including the bacterial chitinases from S. marsecens, B. circulans WL-12 and Alteromonas sp. A. californica nuclear polyhedrosis viral (AcMNPV) chitinase; and the fungal chitinase from Aeromonas caviae. The former two chitinases also have been studied by site-directed mutagenesis and characterization of the kinetic parameters of the purified enzymes. These studies have confirmed that the glutamate in the conserved motif FDGLDLDWEYP is most critical for catalysis and that the aspartate residues play less critical but nonetheless important roles. In this study, we have explored in detail the functional roles of the glutamate and the two aspartic acid residues closest to this glutamate in the linear amino acid sequence of M. sexta chitinase by a series of site-directed mutations. The use of the baculovirus-insect cell line expression system has permitted us to isolate rather large amounts of each of the mutant proteins free of contamination from wild-type or viral chitinases. The endogenous AcMNPV chitinase has a mass of 60 kDa and is not secreted into the medium. The mutant proteins have a mass of approximately 85 kDa and are secreted from the cells.
PKOV is a temperature-sensitive plasmid vector with chloramphenicol resistance and sensitivity of sucrose [37]. We selected the bacterial strains lost plasmid pKOV gradually during passing train at different temperatures with sucrose in medium, and gene replacement occurs when the homologous recombination by RecA protein [38]. Through replacing the 720bp ompR of the bacterial genome by 33bp trailing fragment formed with primer, then we constructed another ompR-deletion mutant strain with the first ompR-deletion used as host strain, the ompR gene mutation fragment (C43→T) recombined to the ompR-deletion genomes, so the 33bp tailing fragment was replaced and the ompR mutant (R15C) was obtained. In this study, rapid screening of the ompR mutant (R15C) was made by the fragments exchange. Pyrobest DNA polymerase was used in this experiment, which is of highly fidelitya in DNA polymerase, and small random mutation rate of the amplified DNA products, the same fidelity performance with Pfμ DNA polymerase and Vent DNA polymerase, as well as the amplification efficiency with Taq DNA polymerase, which is suitable for site-directed mutation.
The method is fast and efficient for vivo site-directed mutation in genome DNA, to avoid adding other marked DNA fragments. General Construction mutant process, it’s difficult to select mutant strains in general process in mutant construction, for pKOV plasmid was often used for the deletion and insertion in large fragment of genome, so we use the fragment exchange, construct genome point mutations step by step, and screened the mutant quickly according to right size of PCR product, so we solve the problem of screening the single base mutation confirmed by sequencing.
Site-directed mutation is an important means to study the relationship between protein structure and function, identification domains or location for specific amino acids. Meanwhile, the technology can also regulate gene expression and DNA molecule (vector) modification to analysis the gene function. The site-directed mutation in vivo can directly transform genes, analysis gene function better [39-40]. With the progress in protein engineering, people will more need to transform their gene products, and the method, site-directed mutation in vivo, is effective for modified genes.
A CKNOWLEDGMENT
The author gratefully acknowledges the financial support from the Education of Zhejiang Province (Y200805545), the Key Fundation of Shaoxing University (08LG1010) and and the Innovation Program of College in Zhengjiang Province (2009).
References Applied an Efficient Site-directed Mutagenesis Method into Escherichia coli
- Kunkel, T.A., Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc. Natl. Acad. Sci. U.S.A. 1985, vol..82, pp. 488–492
- Kunkel, T.A., Bebenek, K., McClary, J., Efficient site-directed mutagenesis using uracil-containing DNA. In: Methods in Enzymology. Academic Press, Inc, 1991, vol.22, pp. 125–139.
- Sugimoto, M., Esaki, N., Tanaka, H., Soda, K., A simple and efficient method for the oligonucleotide-directed mutagenesis using plasmid DNA template and phosphorothioate-modified nucleotide. Anal. Biochem. 1989, .vol.179, pp. 309–311.
- Silaty, S.N., Fung, M., Shen, S.H., Lebel, S., Site-directed mutagenesis by coomplementary-strand synthesis using a closing oligonucleotide and double-stranded DNA templates. Anal. Biochem. 1990, vol.185, pp. 194–200.
- Lewis, M.K., Thompson, D.V., Efficient site directed in vitro mutagenesis using ampicillin selection. Nucl. Acids Res. 1990, vol.18, pp. 3439–3443.
- Deng, W.P., Nickoloff, J.A., Site-directed mutagenesis of virtually any plasmid by eliminating a unique site. Anal. Biochem. 1992, vol.200, pp. 81–88.
- Wong, F., Komaromy, M., Site-directed mutagenesis using thermostable enzymes. BioTechniques, 1995, vol.18, pp. 19-25.
- Ohmori, H., A new method for strand discrimination in sequencedirected mutagenesis. Nucl. Acids Res., 1994, vol.22, pp. 884–885.
- Li, F., Liu, S.L., Mullins, J.I., Site-directed mutagenesis using uracilcontaining double-stranded DNA templates and DpnI digestion. Biotechniques, 1999, vol.27, pp. 734–738.
- Xin, W., Huang, D.W., Zhang, Y.M., Geng, L., DNA mutagenesis using T4 DNA polymerase and DpnI restriction endonuclease. Anal. Biochem. 2004, vol.329, pp. 151–153.
- Lesley, S.A., Bohnsack, R.N., Site-directed mutagenesis using the altered sites II systems. Promega Notes, Promega Corporation, Madison, WI. , 1994, vol.46, pp. 6-10.
- Weiner, M.P., Costa, G.L., Schoettlin, W., Cline, J., Mathur, E., Bauer, J.C., Site-directed mutagenesis of double-stranded DNA by the polymerase chain reaction. Gene, 1994, vol.151, pp. 119–123.
- Lu, L., Patel, H., Bissler, J.J.,. Optimizing DpnI digestion conditions to detect replicated DNA. Biotechniques, 2002, vol.33, pp. 316–318.
- Chiu, J., March, P.E., Lee, R., Tillett, D., Site-directed, ligaseindependent mutagenesis (SLIM): a single-tube methodology approaching 100% efficiency in 4 h. Nucl. Acids Res. 2004, vol.32, pp. 174-178.
- Seyfang, A., Jin, J.H., Multiple site-directed mutagenesis of more than 10 sites simultaneously and in a single round. Anal. Biochem. 2004, vol.324, pp. 285–291.
- Weiner, M.P., Costa, G.L., Schoettlin, W., Cline, J., Mathur, E., Bauer, J.C., Site-directed mutagenesis of double-stranded DNA by the polymerase chain reaction. Gene , 1994, vol.151, pp. 119–123.
- Wu, W., Jia, Z., Liu, P., Xie, Z., Wei, Q., A novel PCR strategy for hight-efficiency, automated site-directed mutagenesis. Nucl. Acids Res. 2005, vol. 33, pp.110-115.
- R.M. Schaaper, R.L. Dunn, B.W. Glickman, Mechanisms of ultraviolet-induced mutation: mutational spectra in the Escherichia coli lacI gene for a wild-type and an excision-repairdeficient strain, J. Mol. Biol. 1987, vol. 198, pp. 187–202.
- R.B. Setlow, W.L. Carrier, F.J. Bollum, Pyrimidine dimers in UV-irradiated poly dI:dC, Proc. Natl. Acad. Sci. USA, 1965, vol. 53, pp. 1111–1118.
- A. Burger, D. Fix, H. Liu, J. Hays, R. Bockrath, In vivo deamination of cytosine-containing cyclobutane pyrimidine dimers in E.coli: a feasible part of UV-mutagenesis, Mutat. Res. , 2003, vol.522, pp. 145–156.
- D. Fix, R. Bockrath, Thermal resistance to photoreactivation of specific mutations potentiated in E. coli B/r ung by ultraviolet light, Mol. Gen. Genet. , 1981, vol.182, pp. 7–11.
- S.K. Banerjee, R.B. Christensen, C.W. Lawrence, J.E. LeClerc, Frequency and spectrum of mutations produced by a single cissyn thymine-thymine cyclobutane dimer in a single-stranded vector, Proc. Natl. Acad. Sci. USA , 1988, vol.85 , pp. 8141–8145.
- S.K. Banerjee, A. Borden, R.B. Christensen, J.E. LeClerc, C.W. Lawrence, SOS-dependent replication past a single trans-syn TT cyclobutane dimer gives a different mutation spectrum and increased error rate compared with replication past this lesion in uninduced cells, J. Bacteriol. , 1990, vol.172, pp. 2105–2112.
- J.E. LeClerc, A. Borden, C.W. Lawrence, The thymine-thymine pyrimidine-pyrimidone(6-4) ultraviolet light photoproduct is highly mutagenic and specifically induces 3_ thymine-to-cytosine transitions in Escherichia coli, Proc. Natl. Acad. Sci. USA , 1991, vol.88, pp. 9685–9689.
- X.D. Zhao, S. Nadji, J.L.F. Kao, J.-S. Taylor, The structure of d(TpA)*, the major photoproduct of thymidylyl-(3-5)-deoxyadenosine, Nucleic Acids Res. , 1996, vol.24, pp. 1554–1560.
- S.N. Bose, R.J.H. Davies, S.K. Sethi, J.A. McCloskey, Formation of an adenine-thymine photoadduct in the deoxydinucleoside monophosphate d(TpA) and in DNA, Science , 1983, vol.220, pp. 723–725.
- S.N. Bose, R.J. Davies, The photoreactivity of T-A sequences in oligodeoxyribonucleotides and DNA, Nucleic Acids Res. , 1984, vol.12, pp. 7903–7914.
- X. Zhao, J.-S. Taylor, Mutation spectra of TA*, the major photoproduct of thymidylyl-(3-5)-deoxyadenosine, in Escherichia coli under SOS conditions, Nucleic Acids Res. , 1996, vol.24, pp. 1561–1565.
- D.F. Fix, N-ethyl-N-nitrosourea-induced mutagenesis in Escherichia coli: multiple roles for UmuC protein, Mutat. Res. , 1993, vol.294, pp. 127–138.
- Steven Forst, Masayori Inouye. Environmentally regulated gene expression for membrane proteins in Escherichia coli. Ann Rev Cell Biol , 1988,vol. 4, pp. 21-42
- Mattison K, Oropeza R, Byers N, et al. A phosphorylation site mutant of OmpR reveals different binding conformations at ompF and ompC.J Mol Biol ,2002, vol.315, pp. 497-511
- Jenny G S, Jamie A. A search for amino acid substitutions that universally activate response regulators.Molecular Microbiology, 2004, vol.51, pp. 887-901
- Nara F, Matsuyama S, Mizuno T, et al. Molecular analysis of mutant ompR genes exhibiting different phenotypes as to osmoregulation of the ompF and ompC genes of Escherichia coli. Mol Gen Genet, 1986, vol.202, pp.194-199
- Takeshi Yoshidal, Ling Qin, Linda A. Transcription Regulation of ompF and ompC by a Single Transcription Factor,OmpR. J Biol Chem, 2006,vol.281, pp.17114-17123
- Li J, Li C, Xiao W, et al.Site-directed mutagenesis by combination of homologous recombination and DpnI digestion of the plasmid template in Escherichia coli. Anaal Biochem,2008, vol.373, pp. 89-391
- Link AJ, Phillips D, Church GM.Methods for generating precise deletions and insertions in the genome of wild-type Escherichia coli:application to open reading frame characterization. J Bacteriol, 1997, vol.179, pp. 6228-6231
- Joseph Sambrook, David W.Russell. Molecular Cloning 3,Huang PT translated,Beijing:science press,2002,pp.96-98
- Hamiliton C M, Aldea M, Washburnet B K, et al.New method for generating deletions and gene replacements in Escherichia coli. J Bacteriology, 1989, vol.171, pp. 4617-4622
- Smith G R.Homologous recombination near and far from DNA breaks: Alternative roles and contrasting views. Annual Review of Genetics, 2001, vol.35, pp. 243-274
- Ling M M and Brian H R. Approaches to DNA Mutagenesis:An Overview.Anal Biochem, 1997, vol.254, pp. 157-178
