Аутовоспалительные заболевания в практике врача-ревматолога. Разбор клинического случая

Бесплатный доступ
Представлено описание аутовоспалительных заболеваний, встречаемых в практике ревматолога, описан клинический случай пациента с аутовоспалительным заболеванием: криопиринассоциированный периодический синдром (САРS), форма: CINCA/NOMID, диагностика и лечение.
Аутовоспалительные заболевания, криопиринассоциированный периодический синдром, ревматология, клинический случай
Короткий адрес: https://sciup.org/170205759
IDR: 170205759 | УДК: 616 | DOI: 10.47475/2409-4102-2024-25-1-74-80
Autoinfl ammatory diseases in the practice of a rheumatologist. Clinical case analysis
The article presents a description of autoinfl ammatory diseases encountered in the practice of a rheumatologist and the development of a patient with an autoinfl ammatory disease: cryopyrin associated chronic syndrome (CAPS), form: CINCA/NOMID, diagnosis and treatment.
Текст научной статьи Аутовоспалительные заболевания в практике врача-ревматолога. Разбор клинического случая
Аутовоспалительные заболевания (АВЗ) — сравнительно новая группа заболеваний. Ген семейной средиземноморской лихорадки был идентифицирован в 1997 г. По своим проявлениям АВЗ могут напоминать известные аутоиммунные заболевания, такие как системная красная волчанка, ревматоидный артрит и т. д. [1; 2].
Теорию и сам термин аутовоспаления ввел в практику Daniel Kastner (1998). Аутовоспаление — патологическое состояние вследствие аномальной активации клеток врожденного иммунитета в результате генетических мутаций, возникает «само по себе» при отсутствии повреждения или инфекции [5; 7].
Центральным механизмом развития многих аутовоспалительных заболеваний является акти- вация инфламмасом. Инфламмасома — белковый комплекс в макрофагах и нейтрофилах, активация которого ведет к секреции провоспалитель-ных цитокинов и запуску воспалительного ответа врожденного иммунитета.
Наследственные периодические лихорадки — группа редких аутовоспалительных заболеваний, встречающихся в практике ревматолога. Эти заболевания связаны с мутациями отдельного гена, ведущими к нарушению регуляции врожденного иммунитета и функции инфламмасом [1; 2].
Следующие заболевания характеризуются повторяющимися приступами фебрильной лихорадки и рецидивирующего системного воспаления с вовлечением многих органов и систем [7]:
-
1. Семейная средиземноморская лихорадка FMF.
-
2. Синдром гипериммуноглобулинемии D HIDS /дефицит мевалонаткиназы МКD.
-
3. Периодический синдром, ассоциированный с рецепторами фактора некроза опухоли-α TRAPS.
-
4. Криопиринассоциированные периодические синдромы CAPS с различной степенью тяжести.
Механизм индукции воспаления при АВЗ может быть различным (рис. 1).
Классификационные критерии Eurofever / PROJECT для наследственных периодических лихорадок разработаны международными экспертами в 2019 г. на основании оценки реальных пациентов из реестра Eurofever. Впервые объединены клинические проявления с генотипом (генетические результаты не являются однозначными, их следует рассматривать в контексте клинических проявлений (%) [11].
Kриопиринассоциированные периодические синдромы (CAPS) представляют собой группу редких аутовоспалительных заболеваний, которая включает [3; 5]:
-
• FCAS семейный холодовой аутовоспалительный синдром;
-
• MWS синдром Макла — Уэллса;
-
• CINCA/NOMID хронический младенческий нервно-кожно-артикулярный синдром / младенческое мультисистемное воспалительное заболевание.
Эти синдромы были первоначально описаны как отдельные нозологические формы, несмотря на некоторые клинические сходства: у пациентов часто присутствуют перекрестные симптомы, включая лихорадку, кожную сыпь, напоминающую крапивницу, и поражение суставов различной степени тяжести, связанное с системным воспалением [1; 2].
Ген, отвечающий за эти три нозологические единицы (FCAS, MWS, CINCA/NOMID), называется NLRP3 и кодирует белок, называемый крио-пирином. Этот белок играет ключевую роль в развитии воспалительной реакции организма. Нарушения в этом гене влекут за собой повышенную функцию указанного белка и повышение воспалительных реакций. Эти усиленные воспалительные реакции вызывают клинические симптомы, наблюдаемые при CAPS.
Клинические проявления и степень тяжести их проявлений во многом схожи, но каждое заболевание имеет свои особенности [6; 7].
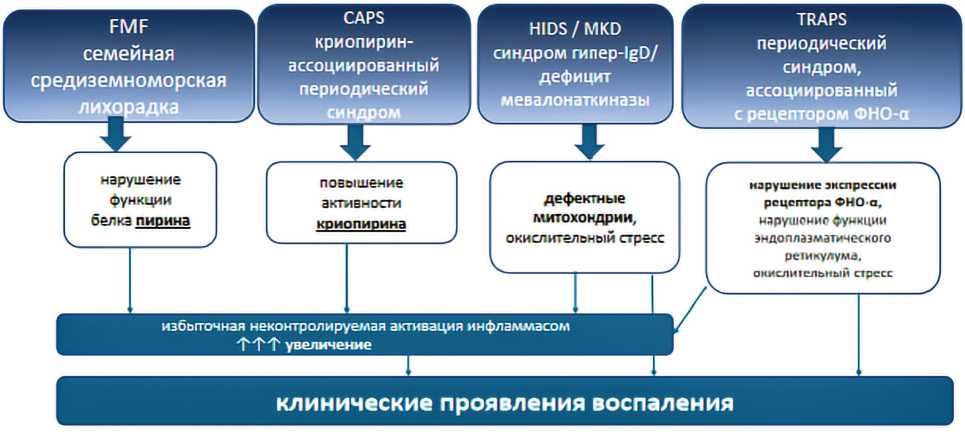
Рис. 1. Механизмы индукции воспаления при семейных наследственных лихорадках [3; 4]
Fig. 1. Mechanisms of inflammation induction in familial hereditary fevers [3; 4]
Грудной возраст
Грудной возраст
С рождения
12-24 ч, после воздействия холода
2-3 дня, часто после воздействия холода
постоянное течение с периодическим усилением
сыпь по типу крапивницы
сыпь по типу крапивницы
нейросенсорная тугоухость; головная боль и асептический менингит;
артралгии
сыпь по типу крапивницы конъюнктивит; головная боль; артралгии хр. менингит; глухота; отек диска зрит.нерва; дисморфизм; задержка интеллектуального развития;
деструктивный артрит
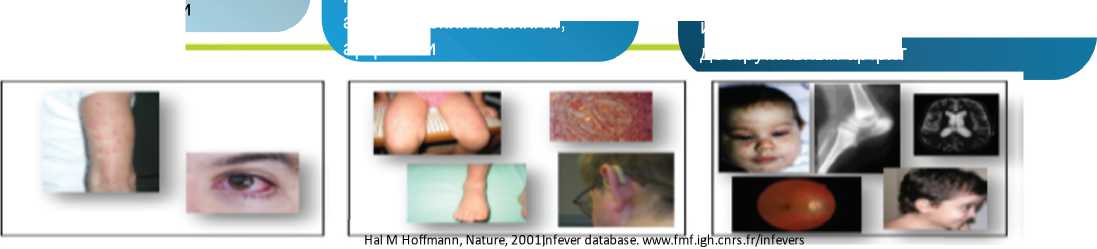
1. Костик ММ и соавт. Как распознать пациента с аутовоспалительным синдромом Современная ревматология 20В;-Ж 14
2. М. Гатторно. Вопросы современной педиатрии. 2014; 13 (2): 5354
3. Салугина СО, Федоров ЕС, Кузьмина НН. Современная ревматология. 2016;10{2)14
Рис. 2. Криопиринассоциированные периодические синдромы (CAPS) с различной степенью тяжести Fig. 2. Cryopyrinassociated periodic syndromes (CAPS) with varying degrees of severity
Наиболее грозным осложнением наследственных аутовоспалительных заболеваний является амилоидоз [8; 9]. В связи с этим своевременная ранняя диагностика АВЗ является актуальной у пациентов детского и взрослого возраста. В ранней диагностике АВЗ основное место принадлежит методам молекулярно-генетической диагностики, которые в настоящее время проводятся в федеральных медицинских учреждениях России.
В Российской Федерации в настоящее время создан проект поддержки пациентов с наследственными аутовоспалительными заболеваниями. Цель данного проекта — поддержка пациентов с наследственными аутовоспалительными заболеваниями (FMF, HIDS/MKD, CAPS, TRAPS) и проведение молекулярно-генетической диагностики в федеральных центрах РФ.
В исследовании могут принять участие пациенты, проживающие на территории РФ, нуждающиеся в верификации диагноза при подозрении на аутовоспалительные заболевания (FMF, HIDS/ MKD, CAPS, TRAPS). Показания к проведению данного вида диагностики при наличии характерных клинических симптомов может устанавливать врач-генетик, врач-ревматолог и в первую очередь врачи-ревматологи, оказывающие специализированную медицинскую помощь по профилю «Ревматология» детскому населению.
В последние годы всё чаще указывается на ведущую роль активации провоспалительного цитокина интерлейкина 1 в патогенезе АВЗ. В кли- нических исследованиях и реальной клинической практике была доказана эффективность ингибиторов интерлейкина 1 (анакинра, канакинумаб) в терапии пациентов с наследственными аутовоспалительными синдромами [10; 11].
В данной статье представлен клинический случай поздней диагностики аутовоспалительного заболевания — ювенильного ревматоидного артрита (ЮРА) с системным началом и криопирин-ассоциированным периодический синдромом (САРS).
Клинический случай
Пациент Х, мужского пола, 1990 г. рождения.
Наблюдается у ревматолога ЧОКБ с августа 2022 г., когда был направлен к ревматологу консультативной поликлиники ГБУЗ ЧОКБ из Магнитогорска с жалобами на снижение зрения на оба глаза, периодические головные боли в височных областях, головокружение. Периодически отмечал повышение температуры до субфебрильнных цифр (до 37,5, 37,2 °С) 1–2 раза в неделю, сопровождающееся чувством жара и общей слабостью. У пациента была выражена сыпь на коже туловища, больше спины и живота, конечностей по типу уртикарной, не сопровождающаяся зудом и болью. На фоне перорального приема лекарственных препаратов отмечал появления болей в эпигастрии. Периодически беспокоило сердцебиение (тахикардия до 100 уд./мин, повышение АД максимально до 135/85–90 мм рт. ст.).
Анамнез заболевания: ребенок родился от первой беременности, в период которой мать перенесла ОРВИ. Роды в срок, оценка по шкале Апгар 7–8 баллов. В возрасте пяти суток кишечное кровотечение (проходил терапию по поводу язвенно-некротического энтероколита, вызванного Е. Colli ), кишечное кровотечение, декомпенсированный дисбактериоз, гипотрофия 2-й ст., перинатальная постгипоксическая энцефалопатия, гипертензионный синдром. В течение 1,5 месяцев получал глюкокортикостероиды (ГКС) — преднизолон в дозе 1,5 мг /кг, в течение пяти месяцев сохранялся неустойчивый стул. В шесть месяцев впервые появилась сыпь на коже. Впервые обратились для дообследования в 1 год 11 месяцев (1992 г.), по месту жительства выставлен диагноз гепатомегалия неясного генеза, хронический активный гепатит. В общем анализе крови (ОАК) умеренный лейкоцитоз до 10–14 · 10 9 , СОЭ 6 мм/ч. Билирубин и трансаминазы в норме, иммуноглобулины А, М, G в норме. В этот же период зафиксированы подъемы температуры тела до фебрильных цифр.
Ходить начал в 12 месяцев.
В 1 год 8 месяцев впервые появилась отечность и боли в суставах, отказывался ходить. В возрасте двух лет направлен к ревматологу, был госпитализирован с симптомами пятнисто-папулезной сыпи, гепатомегалии, артрита коленных суставов, лихорадки. Выставлен диагноз ЮРА (синдром Стилла). Проводилась пульс-терапия метилпреднизолоном и циклофосфамидом, получал индометацин. Выписан на дозе преднизолона 15 мг/сут., на фоне терапии купирован суставной синдром, сыпь купирована на непродолжительный период, затем вновь возникала, но не приносила дискомфорта.
Отмечалась задержка роста. В возрасте пяти лет (1995 г.) впервые направлен в детскую областную поликлинику к эндокринологу в г. Челябинск (рост 97 см, вес 14 кг при поступлении), голова гидроцефальной формы, гипертрихоз, деформация коленных суставов, атрофии мышц нижних конечностей, психическое развитие в норме. По внутренним органам изменений не было выявлено. Заключение эндокринолога — задержка роста и гипертрихоз на фоне приема стероидных препаратов. Пациент продолжает прием глюкокортикостероидов и наблюдается ревматологом амбулаторно.
В семь лет госпитализирован в кардиоревматоло-гическое отделение детской областной больницы.
Проведено полное дообследование и консилиум. Вследствие отсутствия поражения мелких суставов деформация коленных суставов за счет разрастания была исключена; с учетом характерного внешнего вида больного, множественных генетических стигм, диспропорционального телосложения, костных деформаций, нарушения роста зубов, гепатомегалии, псевдозастойного соска зрительного нерва, больше данных за наследственное нарушение обмена (мукополисахаридоз?, остеохондродисплазия?), но уровень гликозаминогликанов в норме.
В 7–8 лет появилось постепенное снижение слуха, использовал слуховой аппарат.
Со слов мамы, в возрасте десяти лет госпитализирован в НИИ педиатрии, где был обследован на I, II тип мукополисахаридоза. Анализы были отрицательные, со слов матери, выставлен диагноз офтальмоостеохондропатия. Рекомендовано снижение дозы ГКС.
При отмене ГКС подъем температуры до фебрильных цифр, ребенок отказывался ходить, вставать. По месту жительства вновь выставлен диагноз ЮРА, к лечению с 2011 г. вновь добавлены ГКС — дипроспан 1 в/м 1 раз в месяц. В 2003– 2004 гг. попытка назначения метотрексата, но из-за развившегося стоматита препарат был отменен. С 2003 г. принимает преднизолон перорально.
В 2005 г. (в возрасте 15 лет) проводился курс плазмафереза трижды, после чего купирована сыпь, начато снижение ГКС. В течение нескольких месяцев чувствовал себя хорошо, затем возвращались явления сыпи, болей в суставах, подъемы температуры тела.
Через три месяца вновь вернулся к приему ГКС 1,5–2 таб./сут., который принимает по настоящее время.
В 2011 г. проведена кохлеарная имплантация.
Наблюдался у ревматолога по месту жительства.
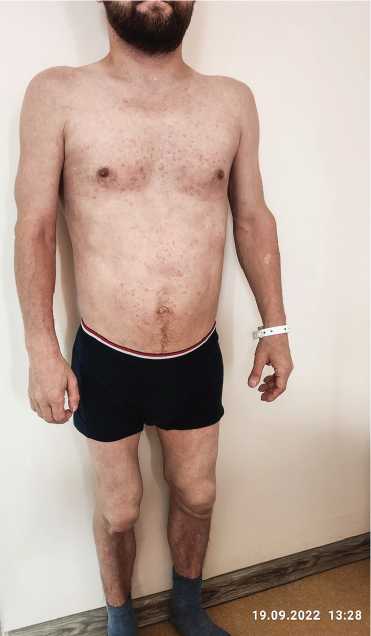
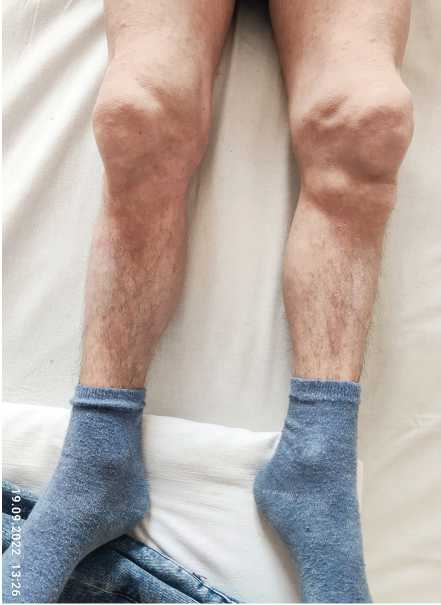
Рис. 3. Низкорослость
Fig. 3. Stunting экстрасистолия 4 а градация по Ryan. Синусовая пароксизмальная тахикардия. Пролапс митрального клапана 1-й степени с регургитацией 1-й степени. Заднекапсулярная катаракта, псевдоза-стойный ДЗН обоих глаз. Стероидный остеопороз. У пациента заподозрено аутовоспалительное заболевание.
Медицинские документы пациента были направлены в ФГБНУ «Научно-исследовательский “Институт ревматологии имени В. А. Насоновой”» в референс-центр по аутовоспалительным заболеваниям. Пациент был госпитализирован для уточнения диагноза и дальнейшей тактики в ФБГНУ «НИИ ревматологии имени В. А. Насоновой»), где проведено обследование, включающее исследование MEF гена, NLRP3, биопсию кожного лоскута, обследование на амилоидоз.
Заключение консилиума: на основании жалоб, лабораторно-инструментальных результатов обследования, осмотра, анализов, анамнеза заболевания у пациента имеется диагноз М08.2 юношеский артрит с системным началом (эритематозная сыпь, субфебрилитет, в анамнезе: артриты, воспалительное заболевание кишечника, гепатосплено-мегалия), криопиринассоциированный периодический синдром (САРS), форма: CINCA/NOMlD
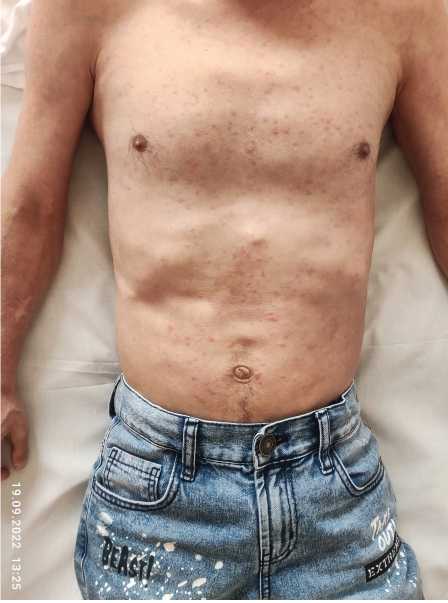
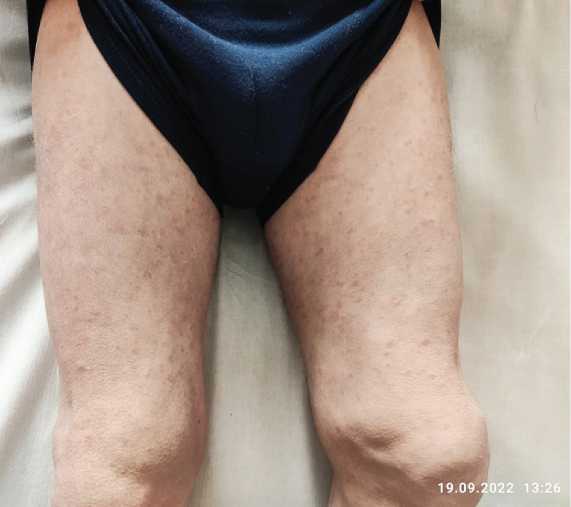
Рис. 4. Типичная сыпь на коже при CAPS
Fig. 4. Typical skin rash with CAPS
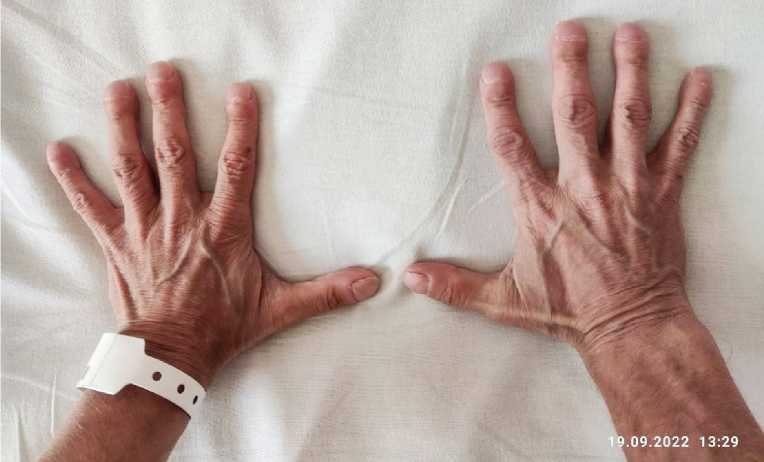
Рис. 5. Изменение суставов
Fig. 5. Joint changes
(младенческий дебют заболевания, крапивница, артралгии, нейросенсорная тугоухость. Ему была назначена и инициирована генно-инженерной биологической терапии (ГИБТ) препаратом из группы ингибиторов ИЛ-1 анакинра (кинерет) 100 мг/сут. подкожно ежедневно в мае 2023 г.
На фоне терапии у пациента полностью купированы симптомы лихорадки, сыпи, нормализовались анализы крови. Пациент продолжает динамическое наблюдение ревматолога и прием препарата кинерет 100 мг/сут.
Выводы
-
• Наследственные периодические лихорадки — редкие, генетически обусловленные хронические заболевания, характеризующиеся высоким статусом системного воспаления.
Прогноз зависит от своевременной постановки диагноза и раннего начала патогенетического лечения.
Благодаря внедрению в клиническую практику молекулярной генетической диагностики, а также лекарственной терапии генно-инженерными биологическими препаратами в выявлении и лечении этих заболеваний достигнут очевидный прогресс.
Пациенты с моногенными периодическими лихорадками встречаются в практике многих специалистов, в связи с чем врачи должны быть готовы к встрече с такими больными.
Идентификация указанных заболеваний основывается на комплексной оценке семейного анамнеза, клинических, лабораторных данных и генетическом тестировании.
Список литературы Аутовоспалительные заболевания в практике врача-ревматолога. Разбор клинического случая
- Кузьмина Н. Н., Салугина С. О., Федоров Е. С. Аутовоспалительные заболевания и синдромы у детей: учеб.-метод. пособие. М.: ИМА-ПРЕСС, 2012. 104 с.: 26 ил.
- Кузьмина Н. Н. Лихорадка неясного генеза в практике педиатра и детского ревматолога // Педиатрия. 2009. No 5. С. 121–127.
- ter Haar NM, Oswald M, Jeyaratnam J и др. Recommendations for the management of autoinflammatory diseases // Annals of the Rheumatic Diseases . 2015 . Vol. 74 , no. 9. P. 1636 – 1644 . DOI: 10.1136/annrheumdis-2015–207546. PMID: 26109736.
- Federici S и др. Frontiers Immunol. 2013. Vol. 4. P. 1–12.
- Georgin-Lavialle S и др. Autoinflammatory diseases: State of the art // Presse medicale. 2019. Vol. 48, no. 1, pt. 2. P. e25-e48.
- Костик М. М., Снегирева Л. С., Дубко М. Ф. и др. Как распознать пациента с аутовоспалительным синдромом // Современная ревматология. 2013. No 3. С. 14–20.
- Салугина С. О., Федоров Е. С., Кузьмина Н. Н. Современные подходы к диагностике, лечению и мониторингу пациентов с криопирин-ассоциированными периодическими синдромами (CAPS) // Современная ревматология. 2016. Т. 10, No 2. С. 4–11. DOI: http://dx.doi.org/10.14412/1996-7012-2016-2-4-11
- Рамеев В. В., Лысенко (Козловская) Л. В., Богданова М. В., Моисеев С. В. Аутовоспалительные заболевания // Клиническая фармакология и терапия. 2020. T. 29, No 4. С. 49–60. DOI: 10.32756/0869-5490-2020-4-49-60.
- Obici L., Merlini G. Amyloidosis in autoinflammatory syndromes // Autoimmunity Reviews. 2012. No 12.С. 14–17.
- Насонов Е. Л. Роль интерлекина 1 в развитии заболеваний человека // Научно-практическая ревматология. 2016. Т. 54, No 1. С. 60–77. DOI: https://doi.org/10.14412/1995-4484-2018-19-27
- Broderick L. Hereditary Autoinflammatory Disorders: Recognition and Treatment // Immunology and allergy clinics of North America. 2019. Vol. 39, no. 1. P. 13–29. DOI: 10.1016/j.iac.2018.08.004. PMID: 30466770.
