Биохимические маркеры метаболизма скелетных мышц, клиническое значение в онкологии: обзор литературы

Автор: Березникова Д.А., Станоевич У.С.
Журнал: Сибирский онкологический журнал @siboncoj
Рубрика: Обзоры
Статья в выпуске: 1 т.25, 2026 года.
Бесплатный доступ
Цель исследования – проанализировать современную литературу о подходах к оценке биохимических маркеров метаболизма скелетных мышц, определить перспективные направления исследований в данной области, обозначить возможные терапевтические стратегии. Материал и методы. Поиск литературы для подготовки обзора осуществлен по базам данных Web of Science, Scopus, Medline, the Cochrane library, РИНЦ, Pubmed. В ходе поиска проанализировано 146 источников, из них в обзор включено 47 публикаций за период 2010–2025 гг. Результаты. Анализ научной литературы позволил выделить 4 ключевых протеолитических каскада, участвующих в развитии саркопении у онкологических пациентов: убиквитин-протеасомный, аутофагический, кальпаин-зависимый и каспазозависимый. Особое внимание уделено прогностической роли биохимических маркеров, включая экспрессию MuRF1 и Atrogin-1, уровни цитокинов и цистатина С. Обнаружена высокая прогностическая значимость соотношения креатинин/цистатин С в оценке риска токсичности противоопухолевой терапии и смертности. Выявлены перспективные молекулярные мишени для таргетной терапии: AMPK, сигнальные каскады IGF-1/AKT/mTOR и транскрипционный фактор NF-kB. Заключение. Саркопения при онкологических заболеваниях обусловлена сложными и взаимосвязанными молекулярными механизмами, включающими как деградацию белка, так и нарушение регенерации мышечной ткани. Использование биохимических маркеров и таргетных вмешательств открывает перспективы для персонализированной диагностики и терапии. Необходимы дальнейшие клинические исследования для валидации биомаркеров и оценки эффективности новых терапевтических стратегий, направленных на предотвращение мышечной атрофии у онкологических больных.
Саркопения, мышечная атрофия, убиквитин-протеасомный путь, аутофагия, кальпаин-зависимая деградация, каспазозависимый путь, маркеры саркопении, терапевтические мишени, протеолитический каскад, кахексия
Короткий адрес: https://sciup.org/140314355
IDR: 140314355 | УДК: 616.74-007.23:616-006 | DOI: 10.21294/1814-4861-2026-25-1-146-154
Biochemical markers of skeletal muscle metabolism and their clinical signifance in oncology: a literature review
Objective: to analyze current approaches to the assessment of biochemical markers of skeletal muscle metabolism, to identify promising research directions in this feld, and to outline potential therapeutic strategies. Material and Methods. The literature search for the preparation of the review was conducted using the Web of Science, Scopus, MEDLINE, Cochrane Library, RSCI, and PubMed databases. A total of 146 sources were analyzed, of which 47 scientifc publications were selected. The review includes studies published between 2010 and 2025. Results. Sarcopenia in cancer patients involves four key proteolytic cascades: the ubiquitin– proteasome pathway, autophagic pathway, calpain-dependent pathway, and caspase-dependent pathway. Particular attention was given to the prognostic role of biochemical markers, including muscle-specifc E3 ligases (MuRF1, Atrogin-1), infammatory cytokines, and cystatin C. A high prognostic value of the creatinineto-cystatin C ratio was demonstrated for assessing the risk of anticancer therapy toxicity and mortality. Promising molecular targets, such as AMPK, IGF-1/AKT/mTOR, and the NF-κB, were identifed for targeted therapy. Conclusion. Sarcopenia in cancer arises from complex and molecular mechanisms, including both protein degradation and impaired muscle tissue regeneration. The use of biochemical markers and targeted interventions opens up prospects for precise, personalized medicine, enabling earlier diagnosis, accurate prognosis, and tailored treatments. Further clinical studies are required to validate biomarkers and evaluate the effectiveness of novel therapeutic strategies aimed at preventing muscle atrophy in cancer patients.
Текст научной статьи Биохимические маркеры метаболизма скелетных мышц, клиническое значение в онкологии: обзор литературы
Изменение метаболизма скелетных мышц играет ключевую роль в патогенезе онкопатологий многих локализаций, особенно в контексте саркопении и раковой кахексии [1–3]. Биохимические маркеры, отражающие метаболическую активность и повреждение мышечной ткани, могут служить важными индикаторами состояния пациента, а также помогают в прогнозировании нежелательных эффектов терапии, исходов лечения.
Среди наиболее изученных показателей особое место занимают миокины (иризин, интерлейкин-6, интерлейкин-15), кахектические факторы (фактор некроза опухоли (TNF-α), интерлейкин-1), протеинмобилизирующий фактор, липид-мобилизирующий фактор, маркеры энергетического метаболизма (лактат, креатинкиназа, аденозинмонофосфат – активируемая протеин-киназа) [4–11]. Ученые отмечают ключевую роль аутофагии в поддержании гомеостаза мышечной ткани [12–16]. В настоящее время публикуется все больше данных о прогностической роли ци-статина С и соотношения кретинин/цистатин С в отношении нежелательных эффектов лечения, клинических исходов [17–19].
Сложная молекулярная структура взаимодействия катаболических и анаболических механизмов, регулирующих гомеостаз скелетных мышц, а также отсутствие единых подходов к оценке и коррекции сниженного мышечного статуса у пациентов с онкопатологией ставят перед клиницистами научно-практическую задачу поиска оптимального алгоритма диагностики, новых терапевтических стратегий в данном направлении.
Цель исследования – проанализировать современную литературу о подходах к оценке биохимических маркеров метаболизма скелетных мышц, определить перспективные направления исследований в данной области, обозначить возможные терапевтические стратегии.
Эволюция понятия « саркопения »
и современные аспекты снижения мышечного статуса у онкологических пациентов
С 1980-х гг. исследователи начали все чаще отмечать, что снижение мышечной массы у пожилых людей оказывает значительное влияние на их подвижность, риск падений и общее качество жизни. Однако для описания этого феномена не существовало четкой терминологии. И. Розенберг предложил термин «саркопения» (от греческих слов «sarx» – мясо и «penia» – утрата или потеря), чтобы акцентировать внимание на утрате мышечной ткани как ключевом аспекте возрастных изменений [20]. Работы И. Розенберга вдохновили множество исследователей в области геронтологии, и в 2010 г. Европейская рабочая группа по саркопении у пожилых людей впервые предложила диагностические критерии саркопении, включающие: оценку мышечной массы, мышечной силы, функциональной производительности [21].
Одним из фундаментальных исследований в области сниженного мышечного статуса онкологических пациентов является работа L. Martin et al. (2013) [1], которые отметили, что саркопения является ключевым предиктором низкой выживаемости, независимо от индекса массы тела пациентов. Исследование С.М. Prado et al. [2] выявило значительные различия в компонентном составе тела, акцентируя внимание на саркопеническом ожирении, где истощение мышц может быть скрыто избыточной жировой массой, что препятствует своевременной диагностике саркопении. Средняя общая выживаемость пациентов с саркопениче-ским ожирением составила 11,3 мес, тогда как у пациентов без саркопении – 21,6 мес (p<0,001). Выявлена тяжелая токсичность (3–4 степени, а именно, лейкопения, нейтропения) у 55 % пациентов с саркопеническим ожирением, по сравнению с 23 % пациентов без саркопении (p=0,004).
В настоящее время опубликованы данные, что не только саркопения, но и миостеатоз является предиктором неблагоприятного прогноза в отношении выживаемости онкологических пациентов. В систематическом обзоре и метаанализе В.К. Лядова и соавт. (2022) [3] подробно рассматриваются методологические аспекты диагностики миостеатоза и его клиническое значение. Авторы анализируют различные диагностические критерии и подчеркивают гетерогенность исследований, связанную с этническими различиями и используемыми пороговыми значениями.
Относительно недавние исследования доказывают, что снижение мышечной массы связано не только с процессом старения и зачастую имеет патологический характер. Патогенез снижения мышечного статуса является мультимодальным процессом, ключевыми факторами которого являются: снижение физической активности, нарушение питания и дефицит белкового компонента, гормональный дисбаланс анаболических и катаболических гормонов, наличие хронических заболеваний и воспалительного процесса, инсулинорезистент-ность. Вышеописанные факторы, взаимодействуя между собой, создают «порочный круг», который ускоряет прогрессирование саркопении [22].
Одной из причин сниженного мышечного статуса в настоящее время принято считать нарушение питания. Европейским сообществом по клиническому питанию и метаболизму в 2017 г. представлена новая классификация, которая включает различные типы недостаточности: саркопения и синдром старческой астении (frailty syndrome), избыточное питание, включая саркопеническое ожирение, недостаточное питание (истощение при заболеваниях и травмах, истощение при голодании, истощение при расстройствах аппетита), нарушение обмена микронутриентов [23, 24].
Патогенетические аспекты саркопении
К вторичной (патологической) саркопении приводят 4 основных механизма, в результате которых происходит усиленная потеря мышечного белка: убиквитин-протеасомный, лизосомально-аутофагальный, кальций-зависимый, каспазозависимый (семья протеолитических энзимов – каспаза-3, М-кальпаин, катепсин L) [25].
Длительное время аутофагия рассматривалась преимущественно как негативная реакция организма на стрессовое состояние, приводящая к катаболизму мышечных волокон и истощению мышечной ткани. Однако в настоящее время выдвигается гипотеза, что клеточная аутофагия является защитным механизмом поддержания состояния скелетных мышц. I. Vanhorebeek et al. [12] рассматривают аутофагию как механизм поддержания клеточного гомеостаза, который играет важную роль в удалении поврежденных органелл, белков и предотвращении апоптоза. Однако в био-птатах мышечной и печеночной ткани критически больных пациентов была выявлена значительно сниженная экспрессия белков, ключевых регуляторов аутофагии. Пациенты с наиболее выраженным подавлением процесса аутофагии имели повышенный риск органной недостаточности и большую частоту летальных исходов. Снижение процесса клеточной аутофагии также коррелировало с худшим функциональным восстановлением у выживших пациентов.
В 2009 г. E. Masiero et al. [13] обнаружили, что при блокировке гена аутофагии (Atg7) наблюдается выраженная атрофия мышц (расширение саркоплазматического ретикулума, появление атипичных гигантских митохондрий, нарушенная организация саркомеров). У мышей с дефектами аутофагии наблюдалось уменьшение площади поперечного сечения мышечных волокон на 20 %, а также значительное снижение силы сокращения (p<0,001). Электронная микроскопия выявила аномальные мембранные структуры и крупные агрегаты белков, положительные на субстраты аутофагии (белок p62, убиквитин). Относительно недавние исследования подтверждают гипотезу, что нарушение аутофагического пути может быть одной из причин возрастной саркопении, миопатий и других заболеваний, сопровождающихся потерей мышечной массы.
I. Vanhorebeek et al. (2011) [14] исследовали влияние нарушения процесса аутофагии у пациентов, длительно находящихся в реанимации. Био-птаты скелетных мышц и печени были получены у пациентов во время плановых хирургических вмешательств (абдоминальных операций) и с последующим использованием методов электронной микроскопии и иммуноблоттинга белков. Целью было определение активности аутофагии по ключевым маркерам LC3-I, LC3-II, p62/SQSTM1 и признакам митофагии. Результаты показали, что у пациентов, несмотря на активацию транскрипционных регуляторов, не происходило должного созревания аутофагосом: отсутствовало увеличение LC3-II, накапливался p62, что указывает на нарушение активации процесса аутофагии. Эти изменения сопровождались накоплением поврежденных митохондрий и белков в клетках печени и мышцах, что, по мнению авторов, усугубляет тканевые повреждения и способствует полиорганной недостаточности.
Относительно недавние исследования показывают, что в клетке существует несколько путей деградации и элиминации клеточных компонентов, основными являются два механизма – процесс аутофагии, играющий роль при длительном воздействии стресс-факторов на клетку; убиквитин-протеасомный, активирующийся преимущественно на ранних этапах стресса [26]. Нарушение процесса аутофагии может стать толчком к развитию сарко-пении. В течение времени в мышечных тканях происходит накопление «дефектных» митохондрий, субстратов клеточного оксидативного стресса, что приводит к повреждению мышечных белков, апоптозу, нарушению регенеративной способности клеток [27, 28].
Процесс аутофагии включает 5 этапов: зарождение фагофора, удлинение его мембраны, захват мишеней, образование аутофагосомы и ее слияние с лизосомой для деградации содержимого. Формирование аутофагосом регулируется комплексом ULK1 (инициирующая аутофагию киназа 1), который активируется AMPK (5’-активированной аде-нозинмонофосфатпротеинкиназой) и подавляется mTORC1 (мишень рапамицина млекопитающих). Белки Beclin-1 (беклин-1) и Vps34 (вакуолярный белок – сортировщик 15/34) способствуют образованию фагофора. Удлинение мембраны обеспечивается убиквитин-подобными системами. Аутофагосомы транспортируются к лизосомам по микротрубочкам, взаимодействуя с моторными белками. Также в процессах аутофагии может участвовать актиновый цитоскелет [15, 16, 29–31].
В настоящее время ключом к разработке терапии, направленной на восстановление мышечной массы и функции, является таргетная коррекция нарушений транскрипционной регуляции аутофагии в скелетной мышце (транскрипционные факторы – FoxO3,TFEB, TFE3, ATF4, NF-kB); использование интервенционных стратегий, способствующих восстановлению метаболического гомеостаза скелетных мышц (применение рапамицина) [32–34]. Несмотря на то, что исследования показывают перспективность модуляции аутофагии и митофагии при мышечной дистрофии, все эти подходы пока остаются на стадии лабораторных или ранних клинических испытаний и не используются в клинической практике.
Синдром анорексии-кахексии
Кахексия – это выраженное истощение (индекс массы тела <17 кг/м2), сопровождающееся снижением мышечного статуса, жирового компонента, которое приводит к полиорганным нарушениям, увеличивая частоту послеоперационных осложнений, влияет на выживаемость и исходы лечения [25]. При наличии вышеописанных состояний, отсутствии снижения индекса массы тела (ИТМТ) менее 17 кг/м2 диагностируется прекахексия. В течении и прогрессировании кахексии выделяют три стадии: прекахексия, выраженная кахексия, необратимая кахексия [25]. Данные патологические состояния выявляются у пациентов с различными злокачественными новообразованиями: опухолями желудочно-кишечного тракта, дыхательной системы, головы и шеи, молочной железы, предстательной железы, кроветворной системы [25, 35]. В основе патогенеза вышеописанного состояния лежит действие цитокиновых (синоним кахектических) факторов (TNF-α, ИЛ-1, ИЛ-6, ИФН-y). Фактор ИЛ-6, лейкемический ингибирующий фактор, онкостатин М активируют сигнальный путь JAK/STAT3, что способствует разрушению мио-фибрильных белков (актин, миозин), стимулирует гормон-чувствительную липазу, вызывая распад триацилглицеролов, увеличивая экспрессию β3-адренергических рецепторов, активируя липолиз и дальнейшие потери жировой ткани. Активация STAT3 приводит к усиленной продукции ИЛ-6, TNF-α, ИЛ-1β, создавая «порочный круг», угнетая анаболические пути [4–6].
Ключевую роль в отношении увеличения мышечной массы играет путь, активируемый инсулиноподобным фактором роста 1 (IGF-1), который связывается с рецептором на поверхности мышечных клеток, что приводит к активации фосфатидилинозитол-3-киназы (PI3K) с последующей активацией Akt (или протеинкиназа B, РКВ). В свою очередь, активация Akt запускает анаболический каскад: стимулирует путь mTORC1, ускоряет транспорт глюкозы в мышечные клетки, блокирует транскрипцию убиквитиновых лигаз [11]. В настоящее время Akt рассматривается как центральный узел регуляции катаболических и анаболических процессов в мышечной ткани. Гиперактивация Akt (например, из-за постоянного стимулирования mTORC1) может быть причиной инсулинрезистентности, митохондриальной дисфункции, раннего «старения» мышечной ткани. Недостаточная активность Akt (при саркопении, раковой кахексии) приводит к ускоренной потере мышечной массы. Ряд исследований показывают, что миостатин ингибирует фосфорилирование Akt, нарушая передачу сигналов через путь IGF-1/ PI3K/Akt. В результате увеличивается уровень активного фактора FoxO, приводя к активации генов, связанных с атрофией [10, 11]. В настоящее время фармакологическая активация Akt рассматривается как стратегия лечения мышечной атрофии и дистрофии.
Также известна роль липидмобилизирующего фактора (ЛМФ) в отношении синдрома анорексии- кахексии. Недавние исследования установили, что ЛМФ является одним из ключевых факторов, ответственных за потерю липидов у онкологических пациентов. ЛМФ увеличивает уровень циклического аденозинмонофосфата в адипоцитах, что приводит к активации гормон-чувствительной липазы и усилению липолиза, усугубляя энергетические потери [39, 40].
Опубликованы данные о роли протеин-мобили-зирующего фактора (ПИФ) в развитии саркопении [7, 41]. Первые исследования этого белка проводили на мышах, данные опубликованы в 1993 г. Были описаны механизмы деградации мышечных белков у животных с экспериментальной моделью кахексии, вызванной аденокарциномой. M.J. Tisdale et al. (2002) опубликовали данные о белке молекулярной массой около 24 кДа, который обладает специфической гликопротеиновой структурой. ПИФ был выделен в крови онкологических пациентов с кахексией и отсутствовал у пациентов без потери массы тела. Современные исследования подтверждают и расширяют гипотезу индуцирующего действия ПИФ на деградацию мышечных белков через убиквитин-протеасомный путь, повышая экспрессию специфических протеасомных субъединиц, усиливая убиквитиновую конъюгацию в мышечных клетках [7, 41].
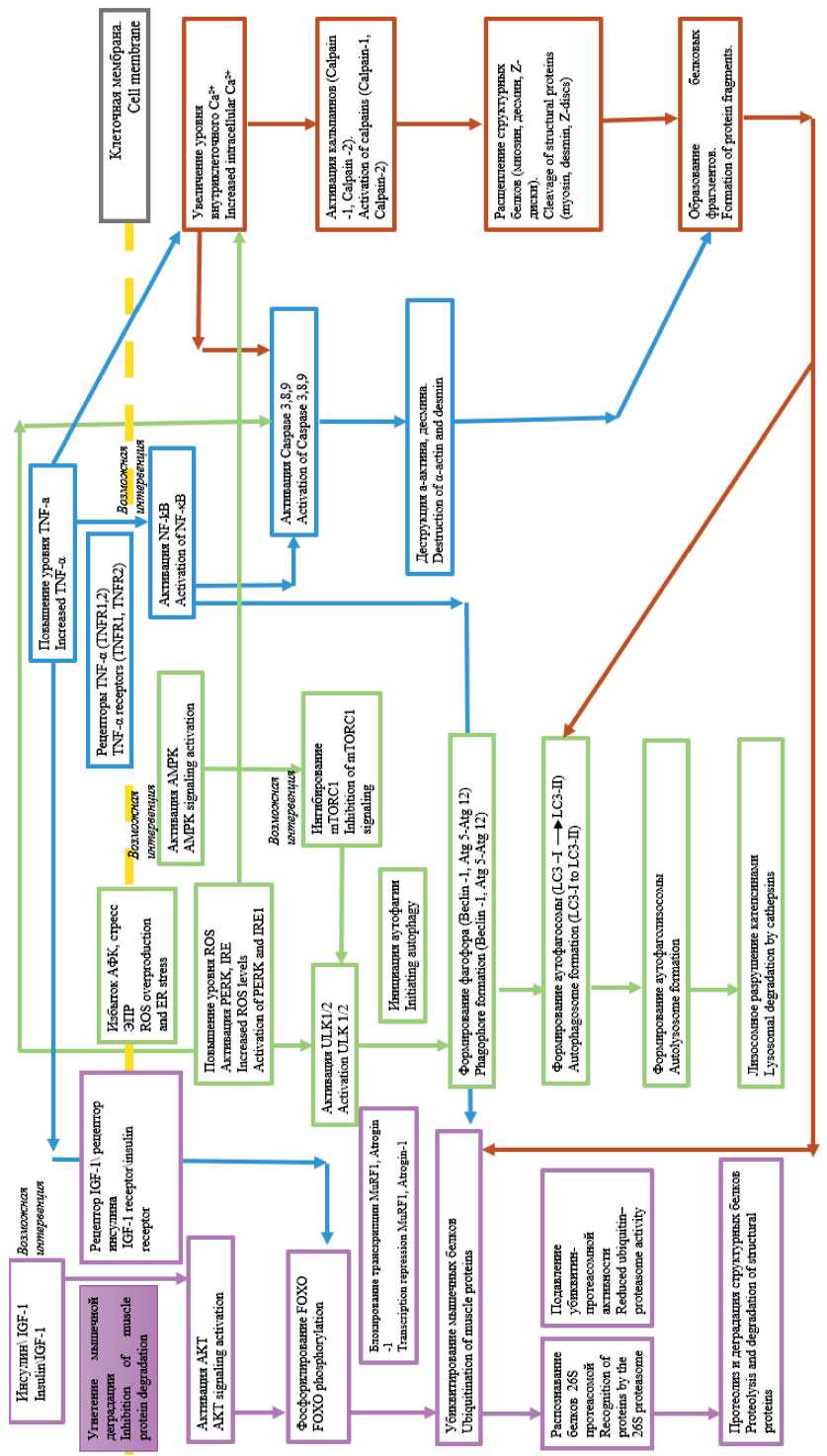
Кальций-зависимый путь деградации мышечного белка играет одну из ключевых ролей в регуляции протеолиза в скелетных мышцах через кальпаины (CAPN1, CAPN2) – (семейство кальций-зависимых цистеиновых протеаз) и связан с развитием кахексии, саркопении и других дегенеративных заболеваний скелетно-мышечной системы. Этот процесс регулируется кальпаино-вым протеолитическим каскадом и взаимодействует с убиквитин-протеасомной системой и процессом аутофагии. Повышенная концентрация внутриклеточного кальция активирует кальпаины, способствуя протеолизу ключевых структурных белков мышечной ткани (десмин, тропонин, тропомиозин, фокальные адгезионные белки), что приводит к ослаблению миофибрилл, нарушению уровня актин-миозинового взаимодействия, дестабилизации сарколеммы. Вышеописанные белки распознаются убиквитиновыми лигазами, метятся убиквитином и утилизируются в ходе убиквитин-протеасомной деградации посредством 26S протеа-сомы, где они разрушаются до коротких пептидных фрагментов. Также кальпаины повреждают ключевые регуляторы аутофагии, способствуя накоплению дефектных белков, снижая регенеративную активность мышечной ткани [8, 42, 43]. Катаболические пути миопатий, возможные терапевтические точки лечения представлены на рис. 1. Убиквитин-протеасомный путь (отмечен фиолетовым цветом) запускается в ответ на воспаление, окислительный стресс и энергетический дефицит (катаболические стимулы). Ключевым звеном является транскрипционный фактор FOXO (центральный транскрипционный фактор), активирующий гены MuRF1 и Atrogin-1, кодирующие убиквитиновые лигазы, которые «помечают» мышечные белки для разрушения. Одновременно энергетический стресс активирует AMPK, который подавляет mTORС1 (мультибелковый комплекс, регулятор синтеза белка, подавления аутофагии) и запускает лизосомально-аутофагический (отмечен зеленым цветом) протеолиз через киназы ULK1/2 (ключевые киназы, инициирующие процесс аутофагии).
При воспалении и стрессе эндоплазматического ретикулума усиливается каспазозависимый путь (отмечен синим цветом): через рецепторы TNF-α активируются каспазы 3, 8 и 9, способствующие апоптозу. Повышение внутриклеточного кальция, характерное для саркопении, активирует кальпаи-ны – протеазы, расщепляющие белки и дополнительно усиливающие апоптоз (кальций-зависимый путь, отмечен коричневым цветом). Данные механизмы функционируют согласованно, взаимно усиливая эффект мышечной атрофии.
Прогностическая роль цистатина С
Цистатин С – низкомолекулярный белок (~13 кДа), который принадлежит к семейству ци-статинов, ингибиторов цистеиновых протеаз. Он играет ключевую роль в регуляции активности цистеиновых протеаз, таких как катепсины B, H и L, которые участвуют в деградации белков, ремоделировании тканей в воспалительных процессах. В нормальных условиях цистатин С регулирует активность катепсинов, предотвращая неконтролируемую деградацию белков. В патологических условиях (воспалительный процесс, онкопатология) дисбаланс между уровнем цистатина С и активностью катепсинов может приводить к разрушению белков внеклеточного матрикса, нарушая тканевый гомеостаз [44].
В настоящее время поступает все больше данных о роли цистатина С и соотношения креатинин/ цистатин С в прогнозировании клинических исходов, токсических осложнений и оценке состояния онкологических больных [17–19, 45]. У пациентов с немелкоклеточным раком легкого, с токсичностью противоопухолевого лечения grade ≥3 выявлено снижение соотношения креатинин/ цистатин С (Cr/CysC). Пациенты с низким соотношением креатинина к цистатину С имели повышенный риск токсичности, особенно при применении платиносодержащих схем химиотерапии [46]. C.Y. Jung et al. [17] опубликовали данные о связи низкого соотношения Cr/CysC с увеличением риска смерти на 34 % (HR=1,34, 95 % CI 1,15–1,58, p<0,001).
Низкий уровень Cr/CysC является прогностическим маркером для пациентов с колоректальным раком. Получены данные, иллюстрирующие отрицательную корреляцию между низким Cr/CysC и

прогрессированием заболевания (PFS) и общей выживаемостью (OS). Показатели PFS у пациентов с низким уровнем Cr/CysC составили 50,8 % против 63,9 % у пациентов с высоким Cr/CysC (p=0,002); OS – 52,5 vs 68,9 % (p<0,001) [18].
Данное отношение коррелирует с онкологическим маркером СА-125 у пациенток с раком яичников (r=0,4, p=0,02 – до химиотерапии; r=0,43, p=0,04 – после первого цикла). Однако не выявлено значительной корреляции между уровнями цистатина C и маркерами скорости клубочковой фильтрации (СКФ). Исследователи пришли к выводу, что цистатин C – ненадежный маркер СКФ при раке яичников из-за его роли как ингибитора цистеиновой протеазы, и его прогностическую роль можно оценивать вне зависимости от функции почек [45].
Обсуждение
Непреднамеренная потеря массы тела (особенно в контексте саркопении, кахексии) является распространенной, нерешенной проблемой в онкологическом сообществе, являясь одним из предиктивных факторов развития нежелательных явлений противоопухолевого лечения, неблагоприятного прогноза. С 1980 г. изучаются аспекты патогенеза данного патологического состояния [7, 12, 13, 20, 39]. В основе различных видов саркопении лежит 4 патогенетических механизма потери мышечной массы, взаимосвязанных между собой: убиквитин-протеасомный, лизосомо-аутофагальный, каспазозависимый и кальпаин-зависимый (кальций-зависимый). Кроме того, свое влияние на прогрессирование данного состояния оказывают такие факторы, как нарушение митохондриального клеточного обмена, оксидативный стресс, стресс эндоплазматического ретикулума, хроническое воспаление.
Найдены терапевтические ключевые точки лечения миопатий: таргетная модуляция транскрипционной регуляции аутофагии в скелетной мышце. Особый интерес представляют транскрипционные факторы, такие как FoxO3, TFEB,
