Биоинформатика и средства компьютерного анализа и визуализации макромолекул

Автор: Порозов Ю.Б.
Журнал: Саратовский научно-медицинский журнал @ssmj
Рубрика: Физиология и патофизиология
Статья в выпуске: 2 т.6, 2010 года.
Бесплатный доступ
В статье представлены цели и задачи биоинформатики. Освещены основные методы и подходы, применяемые в вычислительной биологии. Показаны области, в которых биоинформатика может значительно облегчить и ускорить работу практического биолога и фармаколога. Рассмотрены как базовые пакеты, так и программные средства для полного, глубокого анализа макромолекул и разработки и моделирования лигандов и центров связывания
Биоинформатика, визуализация, макромолекулы, протеомика
Короткий адрес: https://sciup.org/14917057
IDR: 14917057
Bioinformatic science and devices for computer analysis and visualization of macromolecules
The goals and objectives of bioinformatic science are presented in the article. The main methods and approaches used in computer biology are highlighted. Areas in which bioinformatic science can greatly facilitate and speed up the work of practical biologist and pharmacologist are revealed. The features of both the basic packages and software devices for complete, thorough analysis of macromolecules and for development and modeling of ligands and binding centers are described
Текст научной статьи Биоинформатика и средства компьютерного анализа и визуализации макромолекул
-
1С о времени открытия пространственной струк туры ДНК и публикации в журнале Nature статьи Ответственный автор – Порозов Юрий Борисович
50124 Италия, Пиза, виа Моруцци 1. Телефон: (+39)0503153542 E-mail: porozov@sns.it , porozov@ifc.cnr.it
«Структура дезоксирибонуклеиновой кислоты» Джеймса Уотсона и Френсиса Крика [1] в 1953 году, современная медицина и биология сделали гигантские шаги к познанию природы человека, в разработке новых методов диагностики и лечения. Повсе-
- местное распространение получили такие методы исследований, как ЯМР, иммунофлюоресцентная микроскопия, PCR (Polymerase Chain Reaction), FRET (Fluorescence Resonance Energy Transfer). В научных исследованиях применяются многочисленные методы молекулярной биологии, позволяющие синтезировать функционально значимые белки для исследований сигнальных и метаболических путей живой клетки, различные методики цитометрии и методы сепарации и поддержания жизнедеятельности культур ткани, в том числе клеток крови и стволовых клеток. В электрофизиологии широко применяется метод «patch clamp» в различных модификациях, позволяющий регистрировать токи небольшой группы рядом расположенных ионных каналов или единичного канала. Большинство из вышеперечисленных методов невозможно без четкого представления о структуре информационных макромолекул (ДНК и РНК), о функции отдельных их частей и о первичной, вторичной, третичной и четвертичной структурах белковых макромолекул, их изменчивости, активности, физических и биологических свойствах.
Биоинформатика (вычислительная биология) – наука, изучающая последовательности нуклеиновых кислот в ДНК/РНК или аминокислот в белках, их эволюцию, закономерности построения вышеназванных макромолекул, взаимосвязь между последовательностью элементов и пространственной структурой макромолекул, ее физическими свойствами и функциями [2, 3]. Согласно определению Европейского Института Биоинформатики (ЕВІ) биоинф о рмат и ка – это применение компьютерных технологий для управления данными биологии и их анализа [4]. Этот определение подразумевает использование компьютеров и информационных технологий для получения, накопления, анализа и хранения биологических данных. Биоинформатика находится на стыке между математикой, компьютерными науками и биологией и широко использует математическое моделирование и вычислительные мощности не только настольных компьютеров, но и многопроцессорных кластерных систем для решения задач, связанных с управлением огромными объемами информации.
Основные направления биоинформатики: эволюция, поиск и аннотация генов в последовательностях, аннотация и расшифровка геномов, экзон-интронные взаимодействия, классификация и характеризация белков, сравнительная геномика и протеомика, вопросы эволюции белков и геномов, филогенез, структурная биология, разработка специализированных пакетов программ и сетевых сервисов, а также некоторые другие.
Важнейший метод, используемый в биоинформатике для анализа последовательностей, – это упорядочивание, выравнивание (alignment). Существуют парное (pairwise alignment) и множественное (multiple sequence alignment, MSA), локальное [5] и глобальное выравнивания [6]. Суть метода состоит в том, что в двух или нескольких последовательностях производится поиск идентичных или похожих участков. Поскольку в процессе эволюции генов могут происходить мутации, вставки или делеции, то допускается добавление в последовательности «пробелов» (gaps) для получения лучшего результата. В результате такого выравнивания можно выявить нуклеотиды или аминокислоты и их группы (мотивы), имеющиеся во всех сравниваемых последовательностях в определенных позициях. Т.е. можно с большой долей вероятности говорить о том, что именно эти «консер- вативные» участки и есть биологически активные сайты в белках, поскольку они не подверглись изменениям в процессе эволюции. С другой стороны, используя данный метод и задавая в качестве параметра поиска определённый профиль (некоторую последовательность нуклеотидов или аминокислот) можно определить родственные протеины или ДНК и тем самым проследить эволюцию изучаемой последовательности.
Анализ результатов множественного выравнивания аминокислотных последовательностей различных белков или участков ДНК, выделенных из разных организмов, может помочь проследить путь этого белка или гена от организма, в котором он проявился впервые и до более поздних видов. Таким образом, применяя MSA, можно ответить на вопрос ы происхождения того или иного вида, а, зная частоту мутаций, можно определить его примерный возраст.
Парное выравнивание аминокислотных последовательностей двух белков, третичная структура одного из которых неизвестна, позволяет сопоставить первичные структуры. При хороших показателях выравнивания можно говорить о гомологии, о сходстве строения и функции исследуемых протеинов и смоделировать пространственную (3D) структуру. При этом установлено, что белки, имеющие не менее 4050% гомологии в аминокислотных последовательностях, будут иметь похожие третичные структуры [7]. Следовательно, можно предполагать, что и функции таких белков могут быть похожими.
Структурная биология белка [8, 9], являясь частью биоинформатики, широко использует её методы и математический аппарат для решения различных задач, связанных с пространственной организацией белка. Доказано, что именно третичная структура протеина определяет его биологические функции, поэтому чрезвычайно важно знать не только последовательность аминокислот, составляющих белок, но и их взаимное расположение в пространстве. На сегодняшний день существует 2 основных экспериментальных способа определения третичной структуры белка – это рентгеноструктурный анализ и ядерно-магнитный резонанс (ЯМР). Полученные в результате этих исследований данные после обработки заносятся в специальные банки данных – PDB, SRS и SRS3D, SCOP, CATH, PFAM и т.д. Оба метода не лишены недостатков и трудностей при выполнении исследования. Биоинформатика использует совершенно другие подходы для предсказания и визуализации третичной структуры белковых молекул [10,11]: поиск и моделирование гомологов (homology modeling), предсказание на основе законов квантовой механики и молекулярной динамики (Ab initio prediction), предсказание вторичной структуры на основе статистических данных, распознавание фолда (fold recognition или threading), и, наконец, выравнивание структуры (structural alignment). Однако эти методы не могут предсказать точной 3D структуры, поскольку основаны на поиске в различных базах данных уже существующих структур гомологов неизвестной молекулы или на статистически достоверных зависимостях между определёнными последовательностями аминокислот и элементами вторичной структуры. Методы молекулярной динамики требуют огромных вычислительных мощностей и многих лет расчетов (AB initio). Наилучшие результаты (80-95%) дает совмещение этих методов.
Биоинформатика может дать ответы на вопросы, связанные с четвертичной структурой белка и его
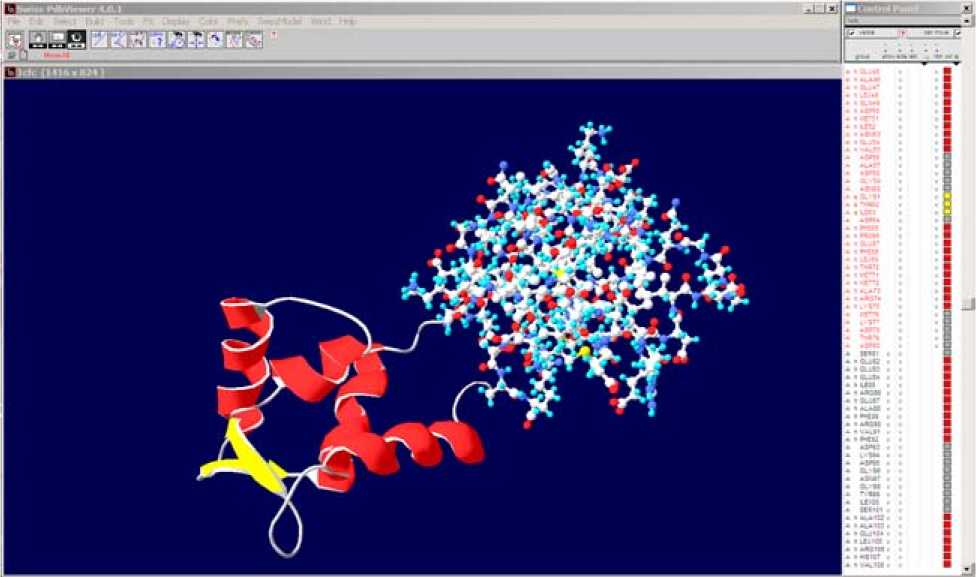
Рис. 1. Окно программы Swiss PDBViewer. Белок кальмодулин (PDB ІD – 1CFC)
взаимодействием с лигандами (docking). Основы ва ясь на 3D структурах взаимодействующих молекул и их физических свойствах (гидрофобность, электростатический заряд, гибкость отдельных цепей, способность к образованию водородных связей) можно предсказать места контакта или связывания этих молекул, рассчитать характеристики связывания. Это позволяет моделировать малые молекулы, способные избирательно активировать или блокировать активные центры целевого протеина. Такое моделирование широко используется в разработке новых лекарств, несмотря на то что требует больших вычислительных и временных ресурсов.
Визуализация и интуитивно понятное представление биологических данных является одной из задач, решаемых в вычислительной биологии. Известно, что до 90% всей информации человек получает от органов зрения, поэтому очень важно, чтобы исследователи обладали инструментарием, позволяющим визуализировать макромолекулы и их комплексы и манипулировать ими.
Для эффективной работы с пространственными структурами белков, их группами, комплексами «белок-ДНК», «белок-лиганд» и т.д. существует специализированное программное обеспечение, позволяющее выбирать нужные структуры из банка данных и отображать исследуемую молекулу или группу в виде, удобном для понимания и анализа. Также многие пакеты имеют средства для создания несложных анимаций, расчета и минимизации потенциальной энергии молекул по одному или нескольким методам и моделирования частей исследуемой молекулы de novo, то есть предоставляют инструментарий для осуществления in silico мутаций, построения петель, реконструкции гомологов, изменения ротамеров радикалов аминокислот, осуществления докинга (предсказание связывания рецептора и лиганда), что чрезвычайно важно при проектировании новых лекарственных средств. На сегодняшний день иссле- дователю доступны как свободно распространяемые программные продукты, так и коммерческие пакеты.
Наиболее известным и бесплатным ПО, в то же время обладающим мощными возможностями для визуализации и моделирования белков, является DeepView-Swiss PDB Viewer (разработчик – the Swiss Іnstitute of Bioinformatics) [12]. Программа позволяет производить базовые операции над данными (рис. 1). В пакете имеется возможность вычислять и минимизировать потенциальную энергию молекулы по методу GROMACS96, выполнять моделирование гомологов (процедура проводится на удалённом сервере SWІSS-MODЕL), строить выравнивание последовательностей аминокислот и производить структурное выравнивание молекул белков. Пользователь может производить базовые операции над полипептидной цепью – строить петли, выполнять мутации, изменять конформации цепи под контролем диаграммы торзионных углов (Ramachandran plot). Имеется также возможность взаимодействовать с этой программой при помощи скриптов, что может позволить автоматизировать рутинные операции.
В распоряжении молекулярных биологов имеются и куда более мощные коммерческие продукты. Пакетом, обладающим огромным потенциалом в области молекулярного моделирования, развитым графическим аппаратом и в то же время позволяющим пользователю сразу начать работу, то есть имеющим дружественный и понятный интерфейс, является Ac-celrys Discovery Studio [13]. Это ПО и само по себе вполне способно удовлетворить большую часть запросов биолога, исследующего белки, но, будучи расширено путём подключения к Accelrys Pipeline Pilot Server, превращается в мощную платформу для моделирования, симуляции и конструирования белков, их комплексов, изучения их взаимодействия в динамике, проектирования белков и проведения QSAR (Quantitative structure-activity relationship – комплекс протоколов, широко используемый при разра-
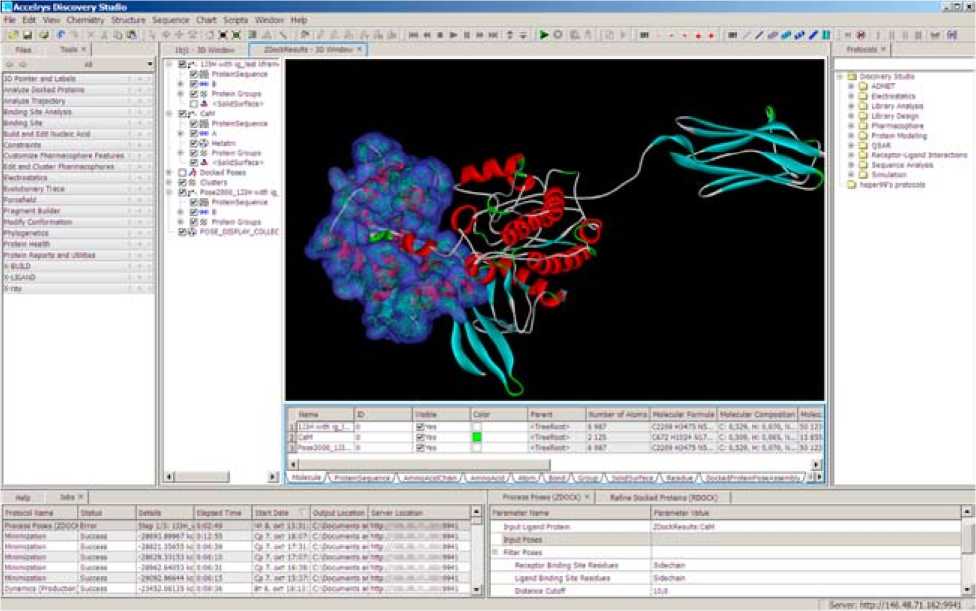
Рис. 2. Окно программ ы Disc o very S t udio. Осуществлён докинг кальмодули н а на MLCK (myos i ne light chain kinase). Ка л ьмодулин связан с α-спиралью кальмодулин-связывающего пептида (Т1711-Т1774). Видны вторичные и третичные структуры, поверхность кальмодулина и интерфейс взаимодействия. Доступные инструменты расположены слева от 3D окна, а протоколы для проведения глубокого изучения взаимодействующих молекул и симуляций – справа. Окна сообщений и вспомогательной информации об исследуемых структурах расположены снизу
ботке новых лекарственных средств). Программный комплекс предоставляет возможность не только изучать различные уровни организации белков, но и осуществлять докинг, исследовать свойства сайтов связывания протеинов, выполнять сложные симуляции при помощи инструментов молекулярной динамики и многое другое (рис. 2).
Серверная часть Discovery Studio открывает доступ к базам данных и инструментам NCBI (National Center for Biotechnology Іnformation), протоколам для исследований в области протеомики, фармакологии, анализа последовательностей и многого другого.
Успехи медицины в настоящее время напрямую зависят от понимания процессов, происходящих на молекулярном уровне. Современные исследования, касающиеся самых актуальных областей, таких как разработка лекарств против вируса иммунодефицита человека и рака, ведутся именно на уровне генов, белков, управляющих транскрипцией, и механизмов регуляции этих процессов. Таким образом, связь биологии, медицины и информационных технологий в ближайшие десятилетия будет укрепляться, поэтому особую важность приобретает проблема преподавания биоинформатики в вузах медико-биологической направленности.
Список литературы Биоинформатика и средства компьютерного анализа и визуализации макромолекул
- Watson J.D., Crick F.H.C. A Structure for Deoxyribose Nucleic Acid//Nature. 1953. V. 171. P. 737-738.
- Attwood T., Parry D. Smith. Introduction to Bioinformatics//Pearson education ltd. 1999. 238 p.
- Lesk A. Introduction to Bioinformatics. Oxford, New York: Oxford University Press, 2008. 474 p.
- The European Bioinformatics Institute. [Electronic resource]. -Electronic data. -Mode access: http://www.ebi. ac.uk/2can/bioinformatics/bioinf_what_1.html
- Smith T.F., Waterman M.S. Identification of common molecular subsequences//J Mol Biol. 1981. №147 (1). Р. 195197.
- Needleman S.B., Wunsch C.D. A general method applicable to the search for similarities in the amino acid sequence of two proteins//J Mol Biol. 1970. №48 (3). Р. 443-453.
- Chothia C., Lesk A.M. The relation between the divergence of sequence and structure in proteins//Embo J. 1986. № 4 (5).Р. 823-826.
- Branden C., Tooze J. Introduction to Protein Structure. Second edition, New York: Garland pub, 1999. 410 p.
- Fersht, A. Structure and Mechanism in Protein Science: a Guide to Enzyme Catalysis and Protein Folding. New York: W.H. Freeman and Co, 1999. 631 p.
- «Computational Methods for Protein Foldings/ed. R.A. Friesner. -Advances in Chemical Physics. 2002. V. 120. 505 p.
- Forster M. Molecular Modelling in Structural Biology//Micron. 2002. V. 33. Р. 365-384.
- Guex N., Peitsch M.C. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling//Electrophoresis. 1997. №18 (15). Р. 2714-2723.
- Accelrys Software Inc. [Electronic resource]. Electronic data. Mode access: http://accelrys.com/
