Bolalarda Alport sindromi va nefrotik sindrom komorbid kechishining immungenetik xususiyatlari

Author: Raxmanova Lola Karimovna, Toshkent tibbiyot akademiyasi, Boltaboeva Muqaddas Mashrabovna
Journal: Re-health journal @re-health
Article in issue: 1 (21), 2024.
Free access
Bolalarda Alport sindromi va nefrotik sindrom komorbid kechishining immungenetik xususiyatlarini oʻrganish maqsadida 3-18 yoshdagi 65 nafar bemor bolalar kuzatuvimizda bo`ldi. Bundan: 35-Nefrotik sindrom (I-guruh); 16-Alport sindromi+Nefrotik sindrom (II-guruh); 14-Alport sindromi (III-guruh). Tadqiqot natijasida ma’lum bo`ldiki, bolalardagi Alport sindromida ikkilamchi nefrotik sindromning qo`shilishi oqibatida (gipoproteinemiya, gipoalbuminemiya, giperxolesterinemiya, giperkoagulyasiya, gipoimmunoglobulinemiya) kasallik progressiyasi va oqsilga bog‘liq boʻlgan ikkilamchi immun etishmovchilik xolatlari (buyrak va oʻpka ABL, IL- 2 ortishi, C3, C4 komponentlari kamayishi, HLA-DR2 antigeni chastotasining sezilarli darajada ortishi) kelib chiqadi. Alport sindromida oqsilga bog‘liq boʻlgan ikkilamchi immun etishmovchilik xolatlari buyrak toʻqimasi shikastlanishi va oʻchoqli segmentar glomeruloskleroz progressiyasida patogenetik jihatdan muxim oʻrin tutadi va oqibatda surunkali buyrak etishmovchiligi rivojlanishini tezlashuviga olib keladi. Kasallikning COL4A3 autosom-retsessiv turida klinik, laborator, immunologik silgishlarning namoyon bo`lishi, glomerulyar filtratsiya tezligining pasayishi, surunkali buyrak yetishmovchiligi rivojlanishi boshqa mutatsiya guruhlariga nisbatan yuqori boʻlib, surunkali buyrak kasalligiga oʻtishining asosiy xavf omilidir. Alport sindromida HLA-DR2 antigeni chastotasining sezilarli darajada ortishi kasallik kechishida oʻchoqli-segmentar glomeruloskleroz progressiyasining yuqori xavf omili bo`lib xisoblanadi.
Immunitet, nefrotik, Alport, sindrom, bola
Short address: https://sciup.org/14130741
IDR: 14130741 | UDC: 616-6
Иммуногенетические особенности коморбидного течения синдрома Альпота и нефротического синдрома у детей
С целью изучения иммуногенетических особенностей коморбидного течения синдрома Альпорта и нефротического синдрома у детей под нашим наблюдением находились 65 детей в возрасте от 3 до 18 лет. Из этого: 35-Нефротический синдром (I группа); 16-синдром Альпорта+нефротический синдром (II группа); 14-синдром Альпорта (III-группа). В результате исследования установлено, что при синдроме Альпорта у детей за счет присоединения вторичного нефротического синдрома (гипопротеинемия, гипоальбуминемия, гиперхолестеринемия, гиперкоагуляция, гипоиммуноглобулинемия) происходит прогрессирование заболевания и случаи белковозависимой вторичной иммунной недостаточности (увеличение АСЛ почек и AСЛ легких, увеличение IL-2, снижение компонентов C3, C4, значительное увеличение частоты антигена HLA-DR2). При синдроме Альпорта белково-зависимый вторичный иммунодефицит имеет патогенетическое значение в прогрессировании поражения почечной ткани и фокально-сегментарного гломерулосклероза и в конечном итоге ускоряет развитие хронической почечной недостаточности. При аутосомно-рецессивном типе заболевания COL4A3 клинико-лабораторные и иммунологические изменения, снижение скорости клубочковой фильтрации, развитие хронической почечной недостаточности выше, чем при других группах мутаций, и являются основным фактором риска перехода в хроническую болезнь почек. Значительное увеличение частоты антигена HLA-DR2 при синдроме Альпорта считается высоким фактором риска прогрессирования фокально-сегментарного гломерулосклероза в течение заболевания.
Immungenetic characteristics of Alport syndrome and nephrotic syndrome comorbidity in children
In order to study the immunogenetic features of the comorbid course of Alport's syndrome and nephrotic syndrome in children, 65 children aged 3-18 years were observed in our clinic. From this: 35-Nephrotic syndrome (group I); 16-Alport syndrome+Nephrotic syndrome (group II); 14- Alport's syndrome (III-group). As a result of the study, it was found that in Alport's syndrome in children, due to the addition of secondary nephrotic syndrome (hypoproteinemia, hypoalbuminemia, hypercholesterolemia, hypercoagulation, hypoimmunoglobulinemia), disease progression and cases of protein-dependent secondary immune deficiency (kidney and lung ABL, IL-2 increase, C3, C4 components decrease, significant increase in frequency of HLA-DR2 antigen). In Alport's syndrome, protein-dependent secondary immunodeficiency is pathogenetically important in the progression of kidney tissue damage and focal segmental glomerulosclerosis, and ultimately accelerates the development of chronic kidney failure. In the COL4A3 autosomal recessive type of the disease, clinical, laboratory, and immunological changes, decrease in glomerular filtration rate, development of chronic kidney failure are higher than in other mutation groups and are the main risk factor for transition to chronic kidney disease. A significant increase in the frequency of the HLA-DR2 antigen in Alport syndrome is considered a high risk factor for the progression of focal-segmental glomerulosclerosis during the course of the disease.
Text of the scientific article Bolalarda Alport sindromi va nefrotik sindrom komorbid kechishining immungenetik xususiyatlari
Muammoning dolzarbligi. Soʻnggi oʻn yilliklar mobaynida nefrologiya amaliyotiga irsiy va klinik membranologiya sohalarini tekshirish usullari keng miqyosda tadbiq etilishi buyrak va siydik chiqarish a’zolari xastaliklarining kelib chiqish sabablari va jarayoni haqidagi tibbiy va ilmiy tushunchalar koʻlamining keskin kengayishi va chuqurlashuviga olib keldi [1-8].
Bu borada olib borilgan kuzatuvlar keyingi yillarda bolalarda buyrak kasalliklarining nozologik tarkibida irsiy va tug’ma hastaliklarning koʻpayib borayotganligini koʻrsatmoqda:14-20% buyrak kasalliklari irsiy omillar bilan bog‘liq tarzda yuzaga keladi va 50% dan ziyod hollarda ularda irsiy moyillik aniqlanadi. Buyrakning irsiy kasalliklari chaqaloqlik davridayoq namoyon boʻlishi mumkin, lekin aksariyat xollarda kech tashxis qoʻyiladi va koʻpincha buyrak faoliyati allaqachon buzilganligi ma’lum boʻladi [9-17].
Tadqiqot maqsadi. Bolalarda Alport sindromi va nefrotik sindrom komorbid kechishining immungenetik xususiyatlarini aniqlash.
Material va usullar. Tadqiqot jarayonida Andijon viloyat bolalar koʻp tarmoqli tibbiyot markazi nefrologiya boʻlimida davolangan 65 nafar bemor bolalar kuzatuvimizda bo`ldi. Bundan: 35-Nefrotik sindrom (I-guruh); 16-Alport sindromi+ Nefrotik sindrom (II-guruh); 14-Alport sindromi (III-guruh). Nazorat guruhi xuddi shu yoshdagi 25 nafar deyarli sog'lom bolalardan iborat bo`ldi.
Quyidagi tekshiruv usullari oʻtkazildi: umumiy klinik tekshiruv (shikoyatlar, anamnezlar, ob’ektiv koʻruv). Oilaviy-irsiy ma’lumotlarni yig‘ish. Geneologik tekshiruv. Qon umumiy va bioximik taxlili. Peshob taxlili. Instrumental tekshiruvlar (UTT buyrak, EKG, audiometriya, vizometriya).
Geneologik usulda bemorlarga shajara tuzildi va shu usul yordamida Alport sindromini qaysi variantga mansubligi (Col4A5, Col4A4, Col4A3) aniqlandi. Alport sindromli (AS) bemorlarning (n=30) 13 nafari I sinf (A va B)va 17 nafari II sinf (DR va DQ) va nazorat guruxidagi sog‘lom bolalarda (n=25) 13 nafari I sinf (A va B) va 12 nafari II sinf (DR va DQ) oʻziga xosliklar sinovdan oʻtkazildi.
va C4 komplement komponentlari immunoturbidimetriya usuli (Z. H. Liu 2003), IL-2 ishlab chiqarilishi immunoferment (IFA) usuli (Aripova T.U., 2004) yordamida aniqlandi [19].
ASni tashxislashda klinik-geneologik usul yordamida kasallikning genetik variantlari aniqlandi. Olingan ma’lumotlar Styudent mezoni qoʻllanilib statistik qayta ishlandi. Oʻrtacha qiymatlar farqlari ahamiyatlilik darajasi P<0,05; P<0,01; P<0,001 boʻlganda ishonchli ma’lumotlar deb qabul qilindi.
Natijalar va muxokama. Tadqiqot natijalarining taxlili shuni koʻrsatdiki, adabiyotlardagi ma’lumotlarni tasdiqlagan holda, AS patogenezining asosi boʻlgan va kasallik debyutida aniqlanuvchi glomerulyar bazal membrana yupqalashuvi va podotsit oyoqchalarining kengayishi sababli filtratsiya jarayonida bazal membranadan eritrotsitlar oʻtib ketishi (gematuriya) keyinchalik proteinuriya va ikkilamchi nefrotik sindrom (NS) rivojlanishi kuzatildi.
Proksimal kanalchalarning epitelial hujayralari tomonidan oqsillar qayta soʻriladi. Bu epitelial hujayralar tomonidan sitokinlar va mediatorlar sintezi va ishlab chiqarilishini kuchaytiradi. Oqibatda tubulointerstitial nefrit rivojlanishi va surunkali buyrak etishmovchiligi (SBYe) progressiyasiga olib keladi.
Tadqiqotlarimiz natijasiga koʻra (1-jadval), ASni ikkilamchi NS bilan komorbid kechishi oqibatida kasallik progressiyasi va NS (gipoproteinemiya, gipoalbuminemiya, giperxolesterinemiya, giperkoagulyatsiya, gipoimmuno- globulinemiya) natijasida oqsilga bog‘liq boʻlgan ikkilamchi immun etishmovchilik xolatlari (buyrak va oʻpka ABL, IL-2 ortishi, C3, C4 komponentlar kamayishi) (P<0,001-0,01-0,05) rivojlandi.
2-jadval ma’lumotlariga koʻra, II va III-guruhlarda IL-2 ishlab chiqarish koʻrsatkichi nazorat guruhiga nisbatan sezilarli darajada ortishi kuzatildi (P<0,001- 0,01-0,05). Ma’lumki, interleykinlar (IL), shu jumladan IL-2 Th1- , Th2 - hujayralari tomonidan sintez qilingan limfokin boʻlib, T-limfotsitlarning koʻpayishi va funksiyasini boshqaradigan asosiy immun boshqaruv ta’sirga ega. Shu bilan birga ushbu sitokin monotsitlar, makrofaglarning oʻsishi, differensiatsiyasi va faollashishiga ta’sir qiladi. Bundan koʻrinadiki, IL-2 ishlab chiqarishdagi buzilish koʻplab patologik jarayonlar, shu jumladan NS rivojlanishida ta’sir koʻrsatishi mumkin.
1-jadval
Tekshirilgan bolalarda antigen bog‘lovchi limfotsit koʻrsatkichlari,M±m
|
Guruhlar |
Qonning ABL,% |
|
|
buyrak ABL, % |
oʻpka ABL,% |
|
|
Nazorat gurux, n =25, 3-18 yosh |
- |
- |
|
NS,n =35, I- guruh |
3,8±0,06* |
0,78±0,064** |
|
AS+NS, n = 16, II– guruh |
4,3 ±0,77** |
0,96 ±0,56*** |
|
AS, n = 14, III –guruh |
0,48 ±0,23* |
0,54 ±0,18* |
Eslatma: * - nazorat guruhga nisbatan ishonchlilik farqini aks ettiradi. *-P<0,001; **-P<0,01; *** P<0,05. ABL I-guruh bilan solishtirgandagi ishonchlilik farqi.
2-jadval
Tekshirilgan bolalarda interleykin-2 koʻrsatkichi, M±m
|
Guruhlar |
Qondagi sitokin, (pg/ml) |
|
IL -2, (pg/ml) |
|
|
Nazorat guruh, n =25, 3-18 yosh |
2,8±0,09 |
|
NS,n =35, I- guruh |
2,4±0,07** |
|
AS+NS, n = 16, II– guruh |
3,0±0,06* # |
|
AS, n = 14, III –guruh |
3,5±0,02*** # |
Eslatma: *-nazorat guruhga nisbatan ishonchlilik farqini aks ettiradi
*P<0,001; **P<0,01; ***P<0,05. #- I, II, III guruhlar oʻrtasidagi ishonchlilik farqi.
3-jadval ma’lumotlari I-chi va II-guruhlarda C3, C4 komplement komponentlari koʻrsatkichlari darajasi kamayishini koʻrsatdi (P<0,001-0,01). Hozirgi vaqtda komplement tizimining C3 komponenti klassik yoʻlda (uning shakllanishi IgG va IgM tomonidan faollashtirilgan) va muqobil yoʻlda ishtirok etishi isbotlangan.
3-jadval
Tekshirilgan bolalarda C3, C4 komponentlar koʻrsatkichi M±m
|
Guruhlar |
Qondagi C3, C4 komponentlar, g/l |
|
|
C3 komponent, g/l |
C4 komponent, g/l |
|
|
Nazorat gurux, n =25, 3-18 yosh |
1,8±0,12 |
0,4±0,13 |
|
NS,n =35, I- guruh |
0,92±0,16** |
0,2±0,18** |
|
AS+NS, n = 16, II– guruh |
1,45±0,13* # |
0,35±0,12* # |
|
AS, n = 14, III –guruh |
1,6±0,16** # |
0,36±0,14** # |
Eslatma: *-nazorat guruhga nisbatan ishonchlilik farqini aks ettiradi.
*P<0,001; **-R<0,01; *** P<0,05. #- I, II, IIIguruhlar oʻrtasidagi ishonchlilik farqi.
C3 komponentining faollashuvi tufayli gistamin semiz hujayralar vatrombotsitlardan ajralib chiqadi, leykotsitlar xemotaksisi va antitanalarning antigen bilan birikmasi, fagotsitoz saqlanadi, tomir devorlarining oʻtkazuvchanligi va silliq mushaklarning qisqarishi kuchayadi. C4 komplement tizimining komponenti faqat fagotsitozni qoʻllab-quvvatlaydigan, qon-tomir devorining oʻtkazuvchanligini oshiradigan va viruslarni zararsizlantirishda ishtirok etadigan komplement tizimini faollashtirishning klassik yoʻlida ishtirok etadi.
Tadqiqotlarimiz natijalari shuni tasdiqladiki, glomerulyar kasalliklarda C3 komplementni tartibga solishning muqobil yoʻli buzilishi muhim rol oʻynaydi. Shu sababli AS NS bilan komorbid kechganda mezangioproliferativ glomerulopatiyaning shakllanishida qon zardobidagi C3, C4 komplement komponenti koʻrsatkichining pasayishi kasallikning og‘ir kechishi va SBYe progressiyasiga sabab boʻlishi mumkinligini tasdiqlaydi. Kuzatuvimizdagi ASli bolalarni klinik-geneologik usul yordamida tekshirilganda 19(63%) nafarida COL4A5 X-xromosomaga bog‘langan turi, 10 (33%) nafarida COL4A3 autosom-retsessiv, 1(4,0%) nafarida COL4A4 autosom-dominant turi qayd qilindi.
Kasallikning COL4A3 autosom-retsessiv turida klinik, laborator va immunologik koʻrsatkichlar, SBYega oʻtishi va glomerulyar filtratsiya tezligining pasayishi boshqa mutatsiya guruhlariga qaraganda yuqori boʻlishi aniqlandi (P<0,001). Bundan koʻrinadiki, ASning aynan autosm-retsessiv (COL4A3) nasllangan turida NSning namoyon boʻlishi fokal-segmentar glomeruloskleroz (FSGS) rivojlanishi va surunkali buyrak kasalligiga (SBK) oʻtishining xavf omili bo`lib xisoblanadi.
Bemorlar va nazorat guruh oʻrtasidagi HLA chastotasini taqqoslash ikki tomonlama Styudent mezoni yordamida baholandi, farqlar P<0,001-0,05 da ishochli deb hisoblandi. Nisbiy xavf (RR), ya`ni HLA antigeni boʻlgan bolalarda kasallik HLA boʻlmaganlarga nisbatan yuqori boʻlishi aniqlandi. Bemorlarda kuzatilgan HLA-A yoki -B antigenlarining chastotasi nazorat guruxidan sezilarli darajada farq qilmadi ( 4-jadval).
HLA-DR2 oʻziga xosligi AS boʻlgan bemorlarda nazorat guruxi bilan taqqoslanganda haddan tashqari koʻp bo`ldi (60%) ( P <0,001). Bemorlarning 18 nafarida HLA-DR2 antigeni ifodalangan, 25nafar nazorat guruxidan 6 nafarida (23%) bir xil antigen mavjud bo`ldi. HLA-DR2 uchun nisbiy xavf va etiologik fraktsiya mos ravishda 5,2 va 0,525 ni tashkil etdi. Bemorlar va nazorat guruhlari oʻrtasida boshqa HLA-DR yoki DQ antigenlarini taqqoslashning statistik tahlili sezilarli farqni koʻrsatmadi (4-jadval).
4-jadval.
Alport sindromli bolalarda HLA turlarining aniqlanish chastotasi M±m
|
HLA antigenlar% P˂0,001 |
|||
|
HLA-A |
HLA-B |
HLA-DR |
HLA-DQ |
|
A 1(50) 14 |
B7(19) 23 |
DR1(29) 30 |
DQ1(65) 53 |
|
A2(52) 50 |
B8(16) 20 |
DR2(23)60 |
DQ2(9) 15 |
|
A3( 20) 4 |
B51(10) 5 |
DR3(25) 17 |
DQ3(44) 17 |
|
A23(6) 3 |
B13(5) 2 |
DR4(32) 37 |
|
|
A24(2) 1 |
B17(6) 5 |
DR5(12) 24 |
|
|
A26( 2) 1 |
B27(3) 2 |
DR6( 13) 12 |
|
|
A11(2) 1 |
B35(13) 13 |
DR7(13) 25 |
|
Eslatma: P˂0,001-nazorat guruhga nisbatan ishonchlilik farqini aks ettiradi.
ASda kuzatilgan ultrastruktura glomerulyar anomaliyalar uchun mas'ul boʻlgan genetik nuqsonlarning xususiyatlari va tabiati xozirgi kunda oʻrganilib kelinmoqda.
Ushbu tadqiqotda biz HLA antigenlari va AS oʻrtasidagi mumkin boʻlgan bog'lanishni baholadik. Bemorlarda kuzatilgan HLA-A yoki -B antigenlarining chastotasi nazorat guruhidan sezilarli darajada farq qilmadi. Tadqiqot natijasiga koʻra, ASli bemorlarda HLA -A yoki -B antigenlari nazorat guruxi bilan bir-xilda ekanligi, bu bilan HLA - A yoki -B antigenlari ASga aloqasi yoʻqligini koʻrsatadi. Aksincha, tadqiqotimizda HLA-DR2 antigeni sezilarli darajada ortdi. Bunga kasallik kechishiga yuqori nisbiy xavf va etiologik fraksion omil sifatida qarash mumkin. Bemorlarimizda kuzatilgan HLA DR7 oʻziga xos chastotasi nazorat guruxidan sezilarli farq qildi va bu mavjud adabiyotlar ma’lumotiga mos keldi. HLA-II sinf antigenlari odatda immunopatogenetik mexanizmlarga ega boʻlgan kasalliklar, masalan revmatoid artrit va 1- tip qandli diabetda uchrashi ma’lum.
AS immunpatogen mexanizmlar bilan kechuvchi kasalliklar guruxiga kirmasada ayrim tadqiqotlarda ASda immunologik anomaliyalar xaqida ma’lumotlar bor.
ASli bemorlarning buyrak biopsiyasi immunofloressent mikroskopik tekshiruvlarida mezangium va glomerulyar kapilyar devorlari boʻylab C3, IgM, IgG va C4 donador choʻkindilari aniqlangan [11,12,13]. Shu bilan birga qon zardobidagi komplementning normal yoki past darajalari, siydikda C3 polipep-tid boʻlaklarining koʻpayishi , plazma va sekretor IgA darajasining pasayishi va allergik kasalliklarning koʻpayishi xaqidagi ma’lumotlar mavjud [4,13].
HLA-si oʻxshash aka-uka oʻrtasida oʻtkazilgan transplantatsiyadan keyin anti-glomerulyar-bazal-membrana (GBM) nefritining paydo boʻlishi IV turdagi kollagen nuqsoni muammosining dolzarbligini tasdiqlaydi [6,8,10]. Boshqa tomondan, anti-GBM nefrit buyrak transplantatsiyasidan oʻtgan ASli erkaklarning 5-10 foizida uchraydi. Patogen anti-GBM antitanalari oddiy glomerulyar va epidermal bazal membranalar bilan ta`sirlanadi lekin ASli bemorlarni aksariyati glomerulyar va epidermal bazal membranalar bilan t`asirlashmaydi. Bundan tashqari, HLA-B7 antigeni HLA-DR2 bilan birgalikda plazmadagi kreatininning yuqori darajasi kasallik prognozini yomonligini koʻrsatuvchi omil xisoblanadi [11,12,13].
MHC (major histocompatibility complex) genlari kollagen sintezi uchun mas'ul boʻlgan genga juda yaqin joylashgan va bu gen, COL11A2, HLA-DP allellariga bir necha kilobaza sentromerikdir [14]. Shuning uchun, HLA II sinf allellari kollagen sintezi uchun mas'ul boʻlgan MHC ichidagi yoki tashqarisidagi boshqa genlar bilan bog'liqlik muvozanatida boʻlishi mumkin. Molekulyar darajadagi qoʻshimcha tahlillar HLADR2 oʻrtasidagi oʻziga xos bog'l anishning asosini tushunishga yordam beradi.
Tadqiqotlarimiz natijalari tasdig‘i sifatida kuzatuvimizda boʻlgan bemorlardan klinik misol keltiramiz.
Bemor Abdusalomov Ozodbek, 14 yosh.
Bemor 9 yil davomida Andijon viloyati bolalar klinikasi nefrologiya boʻlimida davriy ravishda davolanib keladi.
Shikoyatlari: bosh og‘rig‘i, umumiy holsizlik, davriy koʻngil aynishi, sutkalik diurezning kamayishi va siydik rangining oʻzgarishi.
Bolada 5 yoshdan boshlab eshitish qobiliyati pasayishi va miyopiya qayd etilgan.
Anamnesis vitae: bola yaqin qarindoshlik nikohidan (ona tomonidan), 2- homiladorlikdan tug‘ilgan. Onaning birinchi homilasi nobud boʻlgan. Homiladorlikda surunkali pielonefrit, oyoq venalari varikoz kengayishi va temir tanqislik anemiyasi II daraja kasalligi bilan dispanser nazoratida turgan.
Bola vaqtiga etib 2800 g og‘irlikda va 50 sm uzunlikda tug‘ilgan. 1 yoshgacha bolaning jismoniy, somatik va ruxiy rivojlanishi me’yorida kechgan. Ota tomonidagi qarindoshlarda irsiy patologiyalar mavjud boʻlib, amakisida "quyon lab" va tug‘ma yurak nuqsoni (Ochiq arterial kanal) qayd etilgan. Bemorning ammasida ikkinchi tipdagi qanli diabet va buyrak polikiztozi borligi aniqlan. Bemorning onasi xozirgi kunda ham surunkali pielonefrit, oyoq venalarining varikoz kengayishi, ikkinchi darajali temir tanqislik anemiyasi tashxisi bilan dispanser nazoratida turadi.
Bemorning otasi zararli odatlarga ega (alkogolizm, chekish), surunkali pankreatit tashxisi bilan dispanser nazoratida turadi.
Anamnesis morbi: Bemorda kasallikning dastlabki belgilari 5 yoshida kuzatilgan va quyidagi tashxis bilan davolangan: Oʻtkir glomerulonefrit, nefritik sindrom, boshlang’ich belgilar davri, buyrak funksiyasi saqlangan. Hamrox: Tugma yurak nuqsoni (Qorinchalar aro toʻsiq nuqsoni). Surunkali tonzillit toksik-allergik shakli. Eshitish qobiliyatining II darajada pasayishi. Tanqislik anemiyasi II daraja. Bemor yuqoridagi tashxis asosida davolangan, lekin samara kuzatilmagan. Bemor bolaligidan tez-tez bronxit, rinit, sinusit kabi yuqori nafas yoʻllari infeksiyasi bilan yiliga 4-6 marta kasallanadi va dispanser nazoratda turadi.
Status preseans: bemor oʻz tengdoshlaridan jismoniy va aqliy rivojlanishdan sezilarli darajada orqada (4-rasm).
Koʻz va koʻkrak soʻrg‘ichlari orasida gipertelorizm, kindikni pastda joylashuvi, sochlarni peshonagacha oʻsib ketishi, yassilangan burun, quloq supralari nuqsoni, qisqa boʻyin, tishlar diastemasi kabi dizembriogenetik stigmlar aniqlandi (1-rasm).
Teri va shilliq pardalari oqish, rangpar. Periferik limfa tugunlari kattalashgan, og‘riqsiz. Oʻpkada vezikulyar nafas, yurak tonlari ritmik, oʻpka arteriyasi soxasida mayin diastolik shovqin aniqlandi, puls – daqiqasiga 110 marta, arterial qon bosimi 120/70. Tili oq karash bilan qoplangan. Qorni yumshoq og‘riqsiz, jigar va taloq kattalashmagan. Buyrak sohasi patologik oʻzgarishsiz. Kun davomidagi siydik miqdori 850ml (kunlik- 350 ml,tunda-500ml, nikturiya), nisbiy zichligi 1006-1010. Pasternatskiy simptomi ikki tomonlama manfiy. Ich kelishi ravon, kuniga ikki marotaba, shakllangan.
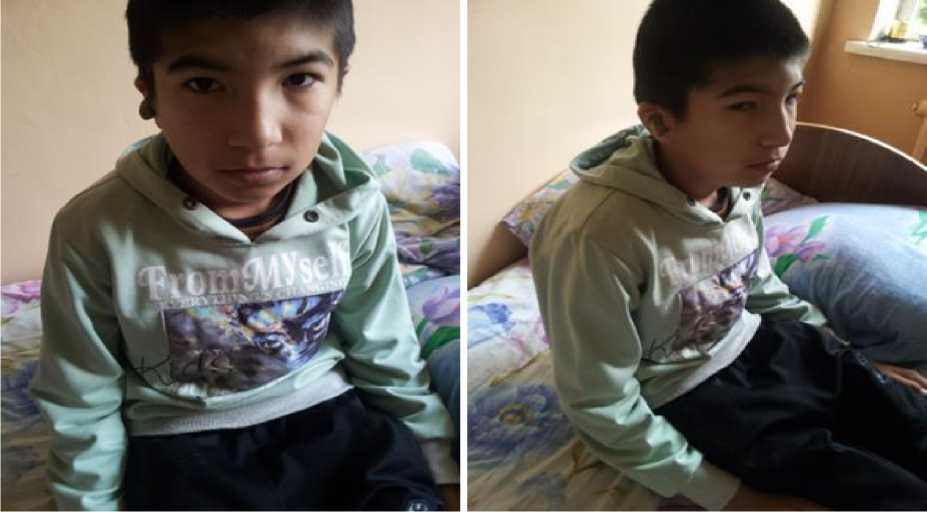
1-rasm . Alport sindromi dizembriogenez stigmlari (sochlarni peshonagacha oʻsib ketishi, yassilangan burun, quloq supralari nuqsoni, qisqa boʻyin).
Laborator taxlillar; umumiy qon taxlili: gemoglobin - 76 g / l, eritrotsitlar - 2,3 x 1012, leykotsitlar - 9,7 x109, ECHT - 18 mm / soat. Umumiy siydik tahlili: miqdori - 50 ml, solishtirma og‘irligi - 1009, siydikda oqsil - 3,0 g/l, yassi epiteliy - 7-9, buyrak epiteliysi- 5-7, leykotsitlar - 3-4, oʻzgargan eritrotsitlar - 6-7, oʻzgarmagan eritrotsitlar -3-4, gialinli silindrlar -3-5. Biokimyoviy qon taxlili: gipoproteinemiya (umumiy oqsil - 42 g/l, gipoalbuminemiya (16%), mochevina - 22,6 mmol/l, kreatinin - 376 mkmol/l, giperxolesterinemiya (14 mmol/l). Immunologik taxlil: ABL-buyrak-4,5%, ABL-oʻpka- 0,96%, IL-2-3,3 pg/ml, C3-1,38/l, C4-0,35/l. HLA-DR2.
Buyraklar ultratovush tekshiruvi: surunkali glomerulonefrit, buyrak parenximasining diffuz oʻzgarishi. Chap buyrak gipoplaziyasi, oʻng buyrak pieloektaziyasi.
LOR tekshiruvi : quloqlarda oʻtkir yallig‘lanish jarayoni yoʻq.
Audiometriya: ikki tomonlama eshitish pasayishi, aralash tipi, III daraja. Oftalmologik tekshiruv: koʻrish oʻtkirligi pasaygan.
Vizometriya: astigmatism II daraja.
Genetik tekshiruv xulosasi : Autosom-retsessiv turdagi nasllanish. Alport sindromining COL4A3varianti aniqlandi.
Klinik, laborator, imminologik, funktsional va genetik tekshiruvlar xulosasiga asosan quyidagi klinik tashxis qoʻyildi:
Irsiy nefrit-Alport sindromi, autosom-retsessiv turi-COL4A3. Asorati: SBYe III-IV bosqich. Nefrotik sindrom. Ikki tomonlama eshitish pasayishi, aralash tipi, III daraja. Astigmatism II daraja Hamrox: Tugma yurak nuqsoni (Qorinchalar aro toʻsiq nuqsoni). Tanqislik anemiyasi oʻrta daraja. Surunkali tonzillit toksik-allergik shakli.
Yuqoridagi ma’lumotlar asosida bemorga konservativ, simptomatik davo va buyrak transplantatsiyasi tavsiya etildi.
Tadqiqot natijalari tasdiqlaydiki, АSli bolalarda HLA-DR2 antigeni chastotasining sezilarli darajada ortishi kasallik kechishida yuqori xavf omili bo`lib xisoblanadi. Ya`ni buyrak translantatsiyasiga tayyorgarlik koʻrayotgan retsipientlarda mavjud boʻlgan HLA antitanalari darajasini aniqlash zaruriy tekshiruv usuli boʻlib, amaliyotda donor buyragini tanlashda muxim rol o`ynaydi va ekstrakorporal gemokorreksiyaga muxtoj boʻlgan bemorlar guruxini aniqlashga imkon beradi. Retsipientga plazmaferez amalga oshirilishi HLA antitanalari kontsentratsiyasi kamayishiga olib keladi va transplantatsiyani muvaffaqiyatli kechish omili hisoblanadi.
Xulosalar:
-
1. Bolalardagi Alport sindromida ikkilamchi nefrotik sindromning qo`shilishi oqibatida (gipoproteinemiya, gipoalbuminemiya, giperxolesterinemiya, giperkoagulyasiya,
-
2. Alport sindromida oqsilga bog‘liq boʻlgan ikkilamchi immun etishmovchilik xolatlari buyrak toʻqimasi shikastlanishi va oʻchoqli-segmentar glomeruloskleroz progressiyasida patogenetik jihatdan muxim oʻrin tutadi va oqibatda surunkali buyrak etishmovchiligi rivojlanishini tezlashuviga olib keladi.
-
3. Kasallikning COL4A3 autosom-retsessiv turida klinik, laborator, immunologik silgishlarning namoyon bo`lishi, glomerulyar filtratsiya tezligining pasayishi, surunkali buyrak yetishmovchiligi rivojlanishi boshqa mutatsiya guruhlariga nisbatan yuqori boʻlib, surunkali buyrak kasalligiga oʻtishining asosiy xavf omilidir.
-
4. Alport sindromida HLA-DR2 antigeni chastotasining sezilarli darajada ortishi kasallik kechishida oʻchoqli segmentar glomeruloskleroz progressiyasining yuqori xavf omili bo`lib xisoblanadi.
gipoimmunoglobulinemiya) kasallik progressiyasi va oqsilga bog‘liq boʻlgan ikkilamchi immun etishmovchilik xolatlari (buyrak va oʻpka ABL, IL-2 ortishi, C3, C4 komponentlari kamayishi, HLA-DR2 antigeni chastotasining sezilarli darajada ortishi) kelib chiqadi.
ADABIYOTLAR:
RHJ-1-2024
References Bolalarda Alport sindromi va nefrotik sindrom komorbid kechishining immungenetik xususiyatlari
- Oʻzbekiston Respublikasi Prezidentining 2018 yil 12 iyuldagi “Oʻzbekiston Respublikasi axolisiga nefrologiya va gemodializ yordami koʻrsatish samaradorligini oshirish chora tadbirlari toʻg‘risida” gi PQ-3846- sonli qarori.
- Axmedov YU.M., Eshqobulov J.E. Bolalar nefro-urologiyasi. Mognografiya. Toshkent – 2021.S-209-218.
- Aksenova ME, Konkova NE, Tutelman KM. Uroven arterialnogo davleniya i progressirovanie patologii pochek u detey s X-sseplennыm sindromom Alporta. Nefrologiya 2020; 24(6):78– 84.(58)
- IgnatovaM.S. Alport-sindrom u detey. Obzor literaturi. Pediatriya 2012; 6: 141—144.
- Papayan A.V., Savenkova N.D. Klinicheskaya nefrologiya detskogo vozrasta –SPb: “Levsha. Sankt-Peterburg”-2008 -600s.
- Poselyugina O.B., Korichkina L.N., Komarova P.M., Troynikova S.A. Klinicheskoe nablyudenie nasledstvennoy nefropatii (sindrom Alporta) //Sovremenniye problemi nauki I obrazovaniya. – 2022. – № 6-1.
- Raxmanova L.K., Boltaboeva M.M.Kliniko-geneticheskie faktori riska razvitiya nasledstvennix nefritov u detey v usloviyax Ferganskoy dolini.RE-HEALTH JOURNAL 2023.№1.1. (17) P.48-55.
- Arellano J, Lehne C, Ojeda S, Sjymansky J, Vasquez L & Kretschmer R (2014). HLA y nefropatias en mestizos mexicanos.Archivos de Investigación Médica , 16:395-400.
- Bekheirnia M.R. i dr. Genotype-phenotype correlation in X-linked Alport syndrome // J. Am. Soc. Nephrol. JASN. - 2010. - № 5 (21). - S. 876-883.
- Burns AP, Fisher M, Li P, Pusey CD Rees AJ (2015). Molecular analyses of HLA class II genes in Goodpasture’s disease. Q uarterly Journal of Medicine , 88:93-100.
- Díez-del Hoyo F, Sanz-Ruiz R, Díez-Villanueva P et al. Anovel cardiovascular presentation of Alport Syndrome: spontaneous coronary artery dissection. IntJ Cardiol 2014;177(3):e133–134
- Gross O., Kashtan C.E., Rheault M.N., et al. Advances and unmet needs in genetic, basic and clinical science in Alport syndrome: report from the 2015 International Workshop on Alport Syndrome. Nephrol Dial Transplant. 2017;32(6):916-924. doi:10.1093/ndt/gfw095
- Kashtan C.E., Ding J., Garosi G., et al. Alport syndrome: a unified classification of genetic disorders of collagen IV α345: a position paper of the Alport Syndrome Classification Working Group. Kidney Int. 2018;93(5):1045-1051.
- Trowsdale J, Ragoussis J & Campbell RD (2001). Map of the human MHC. Immunology Today, 12: 445-446.
- Каримджанов Илхамджан Асамович, Рахманова Лола Каримовна. Некоторые аспекты течения и лечения хронической болезни почек у детей. Журнал Детская медицина Северо- Запада. 2018;7(1):144.
- Rakhmanova Lola Karimovna, Savenkova Nadejda Dmitrievna, Iskandarova Iroda Rustamovna. Immune-hematologikal risks of chronic kidney disease in children with lymphatic diathesis. Журнал Natural Science Edition. 2020;16(10):297-311.
- Rakhmanova Lola Karimovna. Contemporary characterıstıcs of alport's syndrome ın chıldren (literature review).2023;1212(12):49-70.
- Гариб Ф.Ю. и др. Клиническая ценность определения АСЛ у больных брюшным тифом и другими заболеваниями. Метод. Рек. Ташкент. 1983.
- Нормативные показатели основных параметров иммунной системы у детей в возрастном аспекте. Арипова Т.У., Умарова А.А., Петрова Т.А., Нуриева Э.И. Метод. рекомендации. Ташкент. 2004. 16 с.
