Causes, signs, prevention of thalassemia (genetic anemia)
")
Author: Bakhshaliyeva A., Seyidova L., Aliyeva Z., Aliyev C.
Journal: Бюллетень науки и практики @bulletennauki
Section: Медицинские науки
Article in issue: 2 т.11, 2025.
Free access
The medical field has revealed many gene diseases to date. One of these diseases is Thalassemia. This scourge was found about 7 thousand years ago.It was discovered for the first time in the countries of the Mediterranean basin.The word thalassemia is derived from the Greek and means Mediterranean anemia. Thalassemia is one of the most common hereditary blood diseases among different genetic diseases and is inherited by both mother and father. Due to the lack of hemoglobin in the body, the function of erythrocytes is impaired.In this case ,erythrocytes are not disc-shaped, but sickle-shaped. Mutations in our genes produce a protein deficiency in the synthesis of hemoglobin, which leads to the occurrence of thalassemia. This protein contained in erythrocytes gives the blood a red color.Hemoglobin originates in the bone marrow, consists of heme and a globilin (protein) chain.. The iron contained in heme has the ability to combine and separate oxygen into itself. Globulin is composed of 2 alpha, 2 betta,delta or gamma chains. The cause of the occurrence of thalassemia is the absence or partial presence of one of the chains as a result of mutation. In thalassemia, if the synthesis of the “α” chain of hemoglobin is disrupted, it is called “α” thalassemia, and if the synthesis of the ”β” chain is disrupted, it is called “β” thalassemia.
Thalassemia, anemia, hemoglobin, mutation, haemorrhage, oxygen, erythropoiesis, siderosis
Short address: https://sciup.org/14131807
IDR: 14131807 | UDC: 616.419 | DOI: 10.33619/2414-2948/111/23
Причины, признаки, профилактика талассемии (генетической анемии)
Медицина выявила множество генных заболеваний на сегодняшний день. Одно из таких заболеваний - талассемия. Это бедствие было обнаружено около 7 тысяч лет назад. Впервые оно было обнаружено в странах Средиземноморского бассейна. Слово талассемия происходит от греческого и означает средиземноморская анемия. Талассемия является одним из самых распространенных наследственных заболеваний крови среди различных генетических заболеваний и наследуется как по матери, так и по отцу. Из-за недостатка гемоглобина в организме нарушается функция эритроцитов. В этом случае эритроциты имеют не дисковидную, а серповидную форму. Мутации в наших генах вызывают дефицит белка в синтезе гемоглобина, что приводит к возникновению талассемии. Этот белок, содержащийся в эритроцитах, придает крови красный цвет. Гемоглобин образуется в костном мозге, состоит из гема и цепи глобилина (белка). Железо, содержащееся в геме, обладает способностью связывать и разделять кислород. Глобулин состоит из 2 альфа, 2 бетта, дельта или гамма цепей. Причиной возникновения талассемии является отсутствие или частичное наличие одной из цепей в результате мутации. При талассемии, если нарушен синтез «α» цепи гемоглобина, это называется «α» талассемией, а если нарушен синтез «β» цепи, это называется «β» талассемией.
Text of the scientific article Causes, signs, prevention of thalassemia (genetic anemia)
Бюллетень науки и практики / Bulletin of Science and Practice
UDC 616.419
As a result of a violation of the hematopoiesis process and coagulation, blood diseases appear.Violation of the hematopoiesis process ultimately leads to a violation of ertropoesis and leukopoiesis,in which I will give detailed information about thalassemia, which is one of the undesirable difficult-to-coorexed forms of anemia caused by impaired ertropoiesis associated with hereditary genetic factors. What is this disease , this trouble?,how does a person get this disease, is it possible to get rid of this disease and how is it possible? For the first time,thalassemia was described in 1925 by American pediatricians Calley and Lee, and then by Italian authors Rietti and Ckeppi. Iton was told that thalassemia was caused by disruption of globin synthesis.
As we know, there is a primitive hemoglobin-HbP in the erythrocytes of the fetus during the first weeks of development of the womb. At the 9th week of fetal development (HbF), fetal hemoglobin is formed, and before birth, HbA (definitive hemoglobin) (hemoglobin of older people) is formed. After the age of 3, fetal hemoglobin is completely replaced by HbA (elderly human hemoglobin).Thus, fetal hemoglobin has a higher sensitivity to O 2 than definitive hemoglobin, as a result of which it forms a saturated compound with it even under conditions of low voltage of O 2 . In the Normal case, hemoglobin is in a state of 3 physiological compounds. When it binds O 2 , it is converted to oxyhemoglobin-HbO 2 .This compound differs from hemoglobin in color, so arterial blood is crimson. O 2 -free hemoglobin is called restored or dezexyhemoglobin.This is the hemoglobin found in venous blood and gives the blood a dark color.In addition, there is a combination of hemoglobin with CO 2 in venous blood — carbohemoglobin .This, on the other hand, transports CO 2 from the tissues to the lungs. In thalassemia, 50-90% of the hemoglobin belonging to adult individuals is replaced by hemoglobin belonging to the fetus.HbF (fetal) is not able to provide tissues with oxygen at the required level.Therefore, hypoxia develops.Hypoxia, on the other hand, leads to increased erythropoiesis .This also leads to increased absorption of iron and siderosis of organs(accumulation of iron).The content of normal erythrocytes and hemoglobin in the blood decreases. On the other hand,hemolysis increases further because the life span of the formed non-normal erythrocytes is short.In order to determine the normal function of the organism, information is contained in the genes of the human cell.Information related to the hemoglobin protein is transported in a gene located on 2 different chromosomes [2].
Discussion and conclusions of the study
The human 16th chromosome has two genes α globunin, while the 11th chromosome has one β-globin gene.Therefore, α globunin has four alleles,while β-globin has two alleles [3]. When these genes mutate, they cannot perform their function properly. The gene is damaged and what it produces is impaired.people with α andß chain damage are considered to have suffered α,β thalassemia.They cannot produce the right hemoglobin,the synthesis of globilin is reduced,weakened or completely absent. people with a chain damage suffer from a thalassemia. In this case, they prepare hemoglobin either only slightly or not at all.Thus ,it becomes clear that the synthesis of the a chain is determined by 2 allele genes(i.e. 4 genes), and the synthesis of the p chain is determined by 2 allele genes ( i.e. 1 gene). a thalassemia has the following forms that differ from each other:
-
1. Small a thalassemia-the disease is caused by the mutation of one of the genes that ensure the synthesis of the a globin chain (the other 3 genes function normally)or two(the other two genes function normally).In people with one a gene mutated ,indicators of peripheral blood composition, as well as the amount of hemoglobin and erythrocytes, are normal.These are considered healthy individuals and are hereditary carriers of the disease.Weak hypochromic microcytic anemia is identified in people who carry two pathological a thalassemia genes.Other clinical signs do not occur.Small a there is no need for drug treatment in thalassemia.
-
2. Intermediate a thalassemia-in this form of thalassemia, only one of the 4 genes from the a globin chain functions normally.That is, the disease is caused by a hereditary mutation of 3 different genes.During intrauterine development, severe anemia occurs, which occurs with an increase in the amount of hemoglobin Barthan in the fetus.However, despite this, intrauterine death does not occur due to the fact that a fairly normal a globilin chain is also synthesized for fetal development.And after birth, such children have a state of anemia, which varies from moderate to severe throughout their lives .HbH postnantal development in its time, it is the majority.They treat this form of the disease in the same way as in large P thalassemia (homozygous P thalassemia).
-
3. Large a thalassemia-in this form of the disease, all 4 genes of the a globilin chainmutation occurs, and they completely lose their functionand in the end, the a globilin chain is not synthesized at all.This also leads to the synthesis of the y(gamma) globin chain in excess .This also leads to the formation of tetramers of hemoglobin, called HB Barta. Tissue respiration is impaired due to the high opacity of hemoglobin to oxygen in Barta. In this case, the fetus develops signs of severe anemia ,heart failure, disseminated edema ,and death from hydropos (accumulation of water in the body cavity of the fetus)occurs [4].
The disease comes in two forms.One of them is called heterozygous.This is the transmission of an unhealthy gene from one of the parents to the child.That is ,the child received an unhealthy gene from one of the parents, and a healthy gene from the other parent.These individuals are carriers of the disease.If an unhealthy gene is passed on to a child from both parents, it is called a homozygous form of the disease.At this time, the child opens his eyes to the world as a patient with severe thalassemia. Depending on the clinical manifestation and severity,there are major types of thalassemia(Thalassemia major) or Kuli disease, minor and minimal(Thalassemia minor sea minimal). Beta thalassemia is the most common genetic blood disease characterized by decreased production or complete absence of beta-globin chains. Even so, the development of endocrine abnormalities associated with beta thalassemia is still present and is more associated with iron overload, chronic anemia and hypoxia [5].
In individuals with small p thalassemia, only one betta thalassemia gene is found to be Heteresygous.B thalassemia is more common in the world.He is considered the bearer of the sign.It does not count as a disease.In this case,there is a mild,sometimes moderately severe degree of microcytic, hypochromic anemia.The level of iron in the blood serum does not change or is slightly elevated.It is diagnosed according to the finding of Target-like erythrocytes in the blood and a slight increase in fetal hemoglobin (4-6%).There is no hemolytic crisis.It is not required to carry out drug therapy(for example, with iron preparations).Carriers of thalassemia symptoms in general they become healthy individuals.But they can pass this pathological gene on to their children if they do not want it and do not know it.There is no need to treat them.However, some of such patients have mild anemia .That is why many do not even imagine that they are carriers of thalassemia.They find out that they are carriers only after the birth of sick children with large thalassemia or accidentally undergo a special hematological examination.The tendency of thalassemia carriers to other diseases also does not increase.Physical and mental weakness is also not formed in them. However, unfortunately, no matter how hard it is tried, it is impossible to get rid of the carrier of thalassemia. Such individuals cannot develop thalacemia from birth until the end of their lives. The worst thing is that thalasemia carriers, if they feel healthy, pass on the symptoms of the disease to their children by inheritance.
Large в thalasemia (it is also often called Culi's disease, since this disease was first described in 1925 by the Amarican physician-pediatrician Thomas Culi.) under homozygous conditions, two mutants develop as a result of obtaining genes from the same parents.According to its course, it is one of the most severe forms of the disease. In homozygous thalasemia, clinical manifestations of the disease begin to manifest at the end of the 1st year of life or at the beginning of the 2nd year. During the first months ,moderate anemia is noticeable, although the child's condition is considered satisfactory. Subsequently , as a result of a decrease in hemoglobin, a noticeable increase in the size of the spleen, yellow and gray skin , bulging cheekbones, narrowed, cleft eye are observed. There are cases when the child has pallor, weakness, impaired appetite, a large belly physically, such children develop poorly. The facial skeleton is fundamentally changing-a square skull ,a saddleshaped nose, the displacement of the teeth is the coincidence of the lower and upper jaw. They also have a low level of resistance to various infections. They are often infected with infectious diseases and lag behind their peers over the years. To treat a child who has suffered major thalasemia, they need a blood transfusion every month for their entire life. However, this in itself can eventually cause other problems in a sick child. So, due to this disease, iron accumulates in the organs of the treated child. This, in turn, has a bad effect on the functioning of internal organs, disrupting their work. As a result of this, it can lead to cardiovascular failure in patients ,liver disease,endocyrin disorders, its removal as a result of enlargement of the spleen. And thalassemia of the third type is called intermedia,it is usually mild.The disease is detected after one year of age.Such patients do not require completely healthy blood as carriers, nor a lot of blood as patients with large tallasemia.They are periodically transfused with blood. Patients are diagnosed with a blood test,hemoglobin electrophoresis, and genetic tests.
Damage to erythropoiesis is the cause of premature destruction of erythrocytes, chronic anemia in patients with thalassemia ( especially in patients with beta thalassemia), bone marrow enlargement.Patients with Beta thalassemia require blood transfusions, starting in childhood and ending in death. Continuously, the combination of red blood cell transfusions and iron chelation therapy in patients is the most easily accessible supportive treatment and greatly prolongs the survival of patients with thalassemia. Since the disease is Hereditary, the most severe or serious form of the disease occurs during the marriage of two carriers.If two thalassemia carriers are married, there is a 25% chance that a new baby will be born sick,25% healthy,and 50% as a carrier of the disease.And from the marriage of two carriers, 3-kind children can be born.
-
1. Carrier like parents.
-
2. Patient
-
3. A completely healthy child can be born.
No one can know the order in which they were born.A medical examination is required to eliminate these fears.It is a genetic disease that stands at the forefront of the nation,it is a challenge.The reason is that people do not have enough knowledge in this area.. If two carriers start a family, an analysis is taken from the fetus of a pregnant woman between the 10th week and the
17th presence or absence of the disease in the fetus is detected.Artificial childbirth is recommended if there is a is the choice of is up to them to is currently considered the greatest we can give society at least one healthy person ,it will be a great is recommended not to enter into a marriage with a close relative so that the child does not come into the world with this disease. Since thalassemia gene carriers are widespread in Azerbaijan, it is very likely that carriers will meet each other when marriage is established.However, during marriage with a close relative, the risk of passing on the thalassemia gene from the common ancestor to their own children is even risk of having the same defective gene in close relatives is 12.5.Therefore, if the decision to start a family with close relatives is interrupted before marriage, it is necessary to consult doctors and determine the presence or absence of defective who do not undergo the examination until marriage can perform it even after of them if it is determined which one is not a carrier of such thalassemia ,the other does not need to be examined. Because even if one parent is a carrier of thalassemia, a child with large thalassemia is not born in the such families, children are 50% likely to be born healthy and 50% as carriers. If the heads of families ,after passing the examination, seek advice from specialists, then they can manage to prevent the birth of a sick child with large thalassemia.However, even if a child with large thalassemia is not born during family marriages, this disease will manifest itself in their grandchildren .
Treatment of patients with large thalassemia is very complicated and exhausting both for the sick child himself and for his family amount spent on prolonging the lives of these patients, whose treatment is very expensive, turns out to be many times larger than the annual earnings of many a year of treatment of a patient with large thalassemia, at least 10 thousand US dollars are children who cannot be fully cured end their lives before they reach the age of 10 at all. For the treatment of a patient with large thalassemia, bone marrow transplantation is used.Therefore, a significant amount is needed. Preventing the birth of a sick child is considered a stronger method of treatment in hereditary diseases in this way. Such a disease occurs in every region of most common are Gabala,Sheki, Ujar, Goychay, Agdash, Agdam, It is found in zagatala, Ganja, Khachmaz, Imishli, Saatli, Lerik, Astara. In general, there are a lot of carriers.According to statistics for 2015,one in 9 people living in Azerbaijan is a carrier of thalassemia, so up to 1 mln of people have been found to be carriers of thalassemia the world, this figure is 250 million.Marriages in this style are very scary.Azerbaijan is one of the countries where thalassemia is widespread and is considered a land anomaly (so, (for the fruit of light consists in all goodness and righteousness and truth).Every year, about 300 children are born with this type of disease in the Republic .
We are faced with such diseases in every district of Azerbaijan. Most of all in Gabala, Sheki, Ujar, Goychay, Agdash, Agdam, Zagatala, Ganja, Khachmaz,Imishli,Saatli,Lerik,Astara .Carriers as a whole are fiercely numerous. In Nakhchivan,in 2015, there were 27 thalassemia patients, of which 14 were women.If there are so many thalassemia patients, then there were enough carriers. And in 2024, 75 people suffer from blood disease.Several of them died .Currently, there are 41 thalassemia patients registered in Nakhchivan Autonomous Republic .Of these ,22 are women and 19 are men. 10 people living in Nakhchivan city.The youngest of them was born in 2018 , and the largest-in 1956.20 years ago, bone marrow tranplantation was performed on one person, but it failed [9].
In 2023, 385 thalassemia patients were registered with the first diagnosis in their loan is amortized for the remainder of its term. It was noted that 190 of them are persons aged 0-17 the end of 2023, the total number of thalassemia patients registered in medical institutions was 4916, and the number of those aged 0-17 was 2361 .
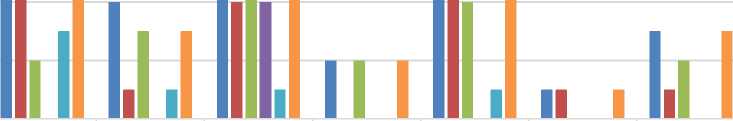
|
0 |
Babek Region |
Ordubad Region |
Nakhchiva n City |
Shahbuz Region |
Sherur Region |
Kengerli Region |
Julfa R |
|
Number of patients |
9 |
4 |
10 |
2 |
10 |
1 |
3 |
|
Female |
7 |
1 |
4 |
0 |
6 |
1 |
1 |
|
Male |
2 |
3 |
7 |
2 |
4 |
0 |
2 |
|
Alpha talassemia |
4 |
||||||
|
Beta talassemia |
3 |
1 |
1 |
1 |
|||
|
Hemolytic anemia D-56 |
7 |
3 |
7 |
2 |
8 |
1 |
3 |
Figure. The number of patients with thalassemia in the 10th international classification of Nakhchivan MR
Currently, bone marrow tranplantation is carried out at the Thalassemia Center in Baku and positive results are achieved. I think that along with a number of diseases, the SOS signal is already being sounded in connection with this is the time to save our gene can be said that the number of people in severe form of the disease does not reach if we pay attention, we will see how many carriers there are for these 1000 children. But I would say that if the population is sufficiently informed and can understand the seriousness of this disease, then at least 10 of them can be he himself is in the hands of this person just need to know a little about it. Many people have no idea about this disease. It is a pity that after their children are born with this disease, they learn that they are carriers of this disease. If they are aware of this disease, if they can understand the seriousness of the disease well, they will be examined before marriage. The result would not be so deplorable either. For parents whose children have severe thalassemia,this is a very serious pain and stress. Basically, the moral blow is very patients should receive treatment for the rest of their becomes for them a mountain of eyes,a mountain of all, what will be the future of such a sick child?, All this must necessarily be communicated to the the same time, children experience great psychological shock. When will all this end?When Will my soul be saved?When Will I grow up?Their integration into society becomes 8-10% of the population is carriers, then the situation is extremely dangerous. In 2015, there were 2,270 thalassemia patients in Azerbaijan, and 40-50% of them needed blood components. according to the information of the Ministry of Health, Shahla Shabanova, a doctor hematologist at the educational - therapeutic Clinic of the Azerbaijan Medical University, said in an interview on May 8, International Thalassemia Day that in 2021, 4,116 thalassemia patients are registered and each of us must take an active part in the education so that this figure does not increase day by are no problems with providing these patients with blood components are determined by the state .
Unfortunately, it is impossible to overcome the moral blow to parents and children. In order to prevent this disease, the government in Azerbaijan has forced couples preparing for marriage to undergo medical examinations since 2015.After that, there was a 45 percent decrease in the number of children born with hereditary pathological blood diseases.This is a significant change.So, before, when couples got married, they did not know that they were carriers of thalassemia, and they did not even have an idea of this disease.Therefore, sick children were born.But after the examination, they realized that they could prevent them from giving birth to a sick child.
According to the Bahadur Eyvazov Research Institute of Hematology, more than half a million people have been examined in Azerbaijan since 2015. The mandatory examination carried out so far from that date revealed 20,000 thalassemia head of the Institute, Zohra Alimirzayeva, said that after learning that both were carriers of thalassemia, about 10 couples changed their minds about marriage .
A law banning the marriage of close relatives ( uncles, aunts, aunts and uncles) has already been approved in Azerbaijan.
In this regard, amendments to the Family Code came into force by Order of President Ilham Aliyev.
With the amendment, the number of cases that prevent the conclusion of marriage is increased.
So, the following circumstances are added to the code. - children of brothers and (or) sisters with a common biological grandfather and (or) grandmother;
The daughter of an uncle (uncle) and a brother (sister), as well as the son of an aunt (aunt) and a brother (sister) with biological kinship.
The law will come into force from July 2025 .
The birth of their children with thalassemia is a serious grief for every parent. During the studies carried out by Azerbaijani hemotologists,it was established that it would take us 50 years to reduce the incidence of thalassemia by 500%.In order to realize this, the marriage of persons with thalassemia should be prohibited. So that, if one of the parties married has thalassemia,then one of the 4 babies born in the family will be doomed to death. If such marriages are prohibited ,then after a generation the result can be successful. In this process, the number of patients who have suffered thalassemia after a generation decreases.In this case, it is necessary to ban 3% of marriages concluded in Azerbaijan. But naturally, this can not be possible.Because who can we limit? To whom do we have the power to create a family with him ?(marry the person we call).Each person must understand this for himself that in families built in this way, unwanted consequences can occur in the end.Therefore, couples should carefully think about their marriage and think about the fate of their future baby in advance .So that, a baby born healthy is a healthy and strong young man of the future. And Healthy Youth means a healthy future. If you want to give birth to a healthy child,if you do not want to regret it for the rest of your life, be sure to get tested before starting a family.If you do not want this network to grow even more,educate those around you so that they can grow socially useful people in them. Since 1993, May 8 has been celebrated annually as international Thalassemia Day.Let's join hands and save our nation ,our future together.To do this ,we need to raise awareness every day, and not on certain days of the year.
References Causes, signs, prevention of thalassemia (genetic anemia)
- Childs B., Valle D. Genetics, biology and disease // Annual review of genomics and human genetics. 2000. V. 1. №1. P. 1-19.
- Nazareth S. A., Əliyev R. A., Əzizov P. P. Genetika və tibb biologiyası kitabı. Bakı, 2008. 828 s.
- Babayev M. A. Genetika tədqiqatları, insanın cəmiyyətdəki rolunun təhlili. Bakı, 2019.
- Carsote M., Vasiliu C., Trandafir A. I., Albu S. E., Dumitrascu M. C., Popa A., Sandru F. New entity - thalassemic endocrine disease: major beta-thalassemia and endocrine involvement // Diagnostics. 2022. V. 12. №8. P. 1921. https://doi.org/10.3390/diagnostics12081921.
- Venou T. M., Barmpageorgopoulou F., Peppa M., Vlachaki E. Endocrinopathies in beta thalassemia: a narrative review // Hormones. 2024. V. 23. №2. P. 205-216. https://doi.org/10.1007/s42000-023-00515-w.
