Дефицит пируватдегидрогеназы у мальчика: клинический случай

Автор: Порецкова Г.Ю., Калинина Е.А., Короткова Н.Н., Болгарова О.Г., Кузнецова Н.Н., Гайсин Ш.И., Бесчастная Е.О.
Журнал: Клиническая практика @clinpractice
Рубрика: Клинические случаи
Статья в выпуске: 2 т.15, 2024 года.
Бесплатный доступ
Обоснование. Дефицит пируватдегидрогеназы - одна из форм тяжёлых наследственных митохондриальных болезней обмена, характеризующаяся нарушениями энергетического обмена и проявляющаяся широким спектром неврологических симптомов. Сложность при подборе терапии заключается в недостаточном объёме информации по ведению детей с данной патологией ввиду их гибели в раннем возрасте и недостаточной диагностики при жизни. Точная распространённость заболевания неизвестна - предположительно, менее 1 случая на 1 000 000, что позволяет отнести его к орфанным заболеваниям. Описание клинического случая. В статье представлен случай наблюдения ребёнка с редким нейрометаболическим заболеванием - дефицитом пируватдегидрогеназного комплекса Е1. Диагноз был заподозрен сразу после рождения на основании неврологической симптоматики, неонатальной гипераммониемии, гиперлактатемии и подтверждён после секвенирования экзома, где выявлен гомозиготный вариант нуклеотидной последовательности в гене PDНА1 (X-19359612-С-Е). В возрасте 2 месяцев ребёнок начал получать кетогенную диету - высокожировую низкоуглеводную сухую смесь для энтерального питания, метаболическую терапию, витамин В1 (по 300 мг/сут), на фоне чего наблюдалась стабилизация клинического состояния. При осмотре в возрасте 9 месяцев отмечена положительная неврологическая динамика, прогноз для жизни на фоне проводимой терапии - благоприятный.
Дети, нейрометаболическое заболевание, мышечная гипотония, неонатальная гипераммониемия, неонатальная гиперлактатемия, дефицит пируватдегидрогеназы, кетогенная диета, тиамин
Короткий адрес: https://sciup.org/143183238
IDR: 143183238 | DOI: 10.17816/clinpract629443
Pyruvate dehydrogenase deficiency in a young boy: a clinical case
BACKGROUND: Pyruvate dehydrogenase deficit is a severe hereditary mitochondrial metabolic disease characterized by impaired energy metabolism and manifested by a wide range of neurological symptoms. The difficulty in selecting therapy is due to insufficient data on the management of children with this pathology owing to death at an early age and insufficient diagnosis during life. The accurate prevalence of the disease is unknown, presumably
Текст научной статьи Дефицит пируватдегидрогеназы у мальчика: клинический случай
Порецкова Г.Ю., Калинина Е.А., Короткова Н.Н., Болгарова О.Г., Кузнецова Н.Н., Гайсин Ш.И., Бесчастная Е.О. Дефицит пируватдегидрогеназы у мальчика: клинический случай. Клиническая прак тика. 2024;15(2):98–105. doi:
Poretskova GYu, Kalinina EA, Korotkova NN, Bolgarova OG, Kuznecova NN, Gaisin SI, Beschastnaya EO. Pyruvate dehydrogenase deficiency in a young boy: a clinical case. Journal of Clinical Practice. 2024; 15(2):98–105. doi:
Submitted 25.03.2024 Revised 07.06.2024 Published online 25.06.2024
из пирувата образуется лактат (молочная кислота), накопление которого в крови и цитоплазме клеток организма вызывает лактатацидоз и, как следствие, изменение ряда метаболических процессов в тканях [5, 6]. Например, избыток лактата запускает активацию клеточных сигнальных путей, которые регулируют воспаление [7], ослабление противоопухолевого иммунитета [8], определяет тяжесть состояния при инфаркте миокарда и кардиогенном шоке [9]. Гликолиз и окисление пирувата происходят уже внутриутробно [10], поэтому дефицит пируватдегидрогеназы, особенно его тяжёлые формы, проявляется сразу после рождения ребёнка, как правило, признаками нарушения функций органов (головной мозг, мышечная ткань), где превращение пирувата в ацетил-КоА происходит наиболее интенсивно [1].
Популяционная частота дефицита пируватдегидрогеназы остаётся неизвестной, тогда как оценки распространённости дефицита пируватдегидроге-назного комплекса варьируют в пределах от 0,9–2,1 (Франция) до 2,7 (Португалия) случаев на 1 млн населения [11]. Дефицит пируватдегидрогеназы возникает в результате изменений генов PDHA1 и PDHB, кодирующих, соответственно, альфа- и бета-субъединицы фермента [4]. Патологические варианты гена PDHA1 являются наиболее частой (до 80% всех случаев) причиной дефицита пируватдегид-рогеназного комплекса. Остальные случаи дефицита вызваны изменениями генов PDHB (4,3%), DLAT, кодирующего E2 (1,5%), DLD, кодирующего E3 (6,2%), PDHX, кодирующего Е3-связывающий белок (10,7%), и PDP1, кодирующего каталитическую субъединицу фосфатазы пируватдегидрогеназы 1, катализирующую фосфорилирование сериновых остатков Е1, что приводит к ингибированию пиру-ватдегидрогеназного комплекса [12, 13].
Наиболее распространёнными клиническими формами дефицита пируватдегидрогеназы являются неонатальная энцефалопатия с лактата-цидозом, ранняя детская форма и форма с поздним началом [14]. Неонатальная форма болезни дебютирует сразу после рождения и отличается высоким риском смерти [6]. Частым клиническим проявлением этой формы болезни является усиливающаяся мышечная гипотония на фоне повышения концентрации лактата в крови [1, 15]. Кроме гипотонии (89%), среди неврологических симптомов отмечают также возникновение мышечной гипертонии или смешанной гипер-/гипотонии (49%), судорог (57%) [1]. Вместе с тем описано относительно доброкачественное течение болезни с мышечной гипотонией и задержкой развития [16]. Неонатальная форма нередко сочетается со структурными аномалиями головного мозга, включая вентрику-ломегалию (67%) и агенезию, дисгенезию или гипоплазию мозолистого тела (55%) [17, 18].
Ген PDHA1 сцеплен с Х-хромосомой, что предопределяет наличие признаков болезни, сцепленной с полом [4], а именно яркую клиническую картину с тяжёлым и среднетяжёлым течением у лиц мужского пола. В ряде описанных примеров подозрение на дефицит пируватдегидрогеназы с последующим обследованием возникает на первом году жизни при наличии у ребёнка сохраняющейся гипотонии, формировании задержки общей и мелкой моторики, развитии судорог [13, 19]. Позднее назначение лечения не позволяет стабилизировать пациента, что приводит к летальному исходу [19].
В литературе приводятся сообщения об использовании дихлорацетата и фенилбутирата как потенциальных методов лечения дефицита пируватдегидрогеназы, вызванного PDHA1 -патогенными вариантами, как временного средства уменьшения лактатацидоза [20].
С учётом механизма развития данной патологии наиболее частыми рекомендациями являются назначение кетогенной диеты с высоким содержанием жиров и низким содержанием углеводов, что позволяет частично уменьшить дефицит пируватдегидрогеназы, но никогда не устраняется полностью [20]. В сообщении K. Sofou и соавт. [21] приводятся данные о снижении уровня лактата крови и некоторых когнитивных улучшениях у пациентов, использующих кетогенную диету более 2 лет.
В статье приведено описание первого в Самарской области клинического случая ребёнка с дефицитом пируватдегидрогеназы. Акцент сделан на необходимости раннего выявления дефицита пиру- ватдегидрогеназы и более раннего начала лечения у новорождённых и детей первых месяцев жизни с необъяснимой прогрессирующей мышечной гипотонией, слабостью, стойкой гиперлактацидемией, отставанием в нервно-психическом развитии.
Поскольку имеются лишь единичные сообщения о раннем назначении терапии детям с такой патологией, представляем опыт лечения ребёнка с назначением ему в возрасте 2 месяцев жизни кето-генной диеты и высоких доз тиамина.
КЛИНИЧЕСКИЙ ПРИМЕР
О пациенте
Мальчик А., 2022 года рождения, декабрь.
Из анамнез а. Родился от второй беременности (первая беременность — нормальные роды в 2021 году, девочка здорова), вторых срочных родов, массой тела 2970 г, длиной 50 см, оценкой по шкале Апгар 7/5 баллов. Беременность протекала на фоне артериальной гипертензии, аутоиммунного тиреоидита, эутиреоза и ожирения II степени. Матери 29 лет, отцу 30 лет; профессиональных вредностей у родителей не выявлено; отец курит; состоят в зарегистрированном браке; наследственных заболеваний не выявлено.
При первом осмотре после рождения состояние оценено как среднетяжёлое за счёт мышечной гипотонии. Состояние ухудшалось за счёт усугубления неврологической симптоматики, в связи с чем в первый день жизни был переведён в отделение реанимации и интенсивной терапии для новорождённых Детской республиканской клинической больницы Республики Татарстан г. Казани (ДРКБ).
Объективно. При поступлении состояние тяжёлое за счёт неврологической симптоматики, метаболических нарушений. Вялый. Глаз не открывает. Рефлексы снижены. По лабораторным данным отмечались гипераммониемия (до 121 мкмоль/л), гиперлактатемия (до 8,1 ммоль/л), повышение общего билирубина за счёт непрямой фракции до 156 мкмоль/л. Уровни исследуемого общего белка, аланинаминотрансферазы, аспартатаминотрансферазы, мочевины, креатинина, калия, натрия в норме. В общем анализе крови воспалительной активности нет, гемоглобин 173 г/л, тромбоциты 381x103. При нейросонографии выявлены дупликационные кисты в сагиттальном срезе, морфофункциональная незрелость. Ультразвуковое исследование гепато-лиенальной системы патологии не выявило. Эхокардиография показала наличие открытого овального окна (3,4 мм) со сбросом слева направо, систолическое давление в лёгочной артерии 26 мм рт.ст., в стволе лёгочной артерии патологических потоков нет. На электроэнцефалограмме зарегистрирована единичная патологическая медленноволновая активность в затылочных областях.
Лечение : ограничена энтеральная белковая нагрузка до 0,5 г/кг, внутривенная дотация глюкозы до 1 г/сут.
Физикальная диагностика
По данным физикального осмотра в первые сутки жизни зафиксировано ухудшение неврологической симптоматики: появились угнетение спонтанной двигательной активности, усиление тремора подбородка, глазодвигательные нарушения в виде нистагма, синдром мышечной гипотонии. Рефлексы снижены.
Предварительный диагноз
При переводе в отделение реанимации выставлен предварительный диагноз: «Перинатальное поражение центральной нервной системы гипок-сически-ишемического генеза в форме синдрома мышечной гипотонии, глазодвигательных нарушений. Неонатальная гипераммониемия, гиперлакта-темия. Наследственные болезни обмена. Метилмалоновая ацидурия. Врождённый порок развития головного мозга — гипоплазия мозолистого тела. Кистовидная трансформация шишковидной железы. Открытое овальное окно. Дополнительная хорда левого желудочка. Ателектаз правого лёгкого. Анемия смешанной этиологии тяжёлой степени».
Динамика и исходы
После стабилизации состояния на 6-й день жизни ребёнок был переведён в отделение патологии новорождённых (ДРКБ: состояние тяжёлое, угнетение спонтанной двигательной активности, усиление тремора подбородка, глазодвигательные нарушения в виде нистагма.
В лечении пациента уменьшено энтеральное питание до 50 мл через 3 часа с допаиванием 5% раствором глюкозы (1,6 г/кг в сутки).
Биологический материал ребёнка отправлен для генетического исследования.
06.02.2023 ребёнок доставлен бригадой санитарной авиации из ДРКБ г. Казани в ГБУЗ СО «Самарская областная клиническая больница имени В.Д. Середавина» (СОКБ им. Середавина) по месту жительства родителей.
Находился в отделении реанимации и интенсивной терапии по 07.04.2023, где с 10.02.2023 по жизненным показаниям начал получать кетогенную диету (лечебная высокожировая низкоуглеводная сухая смесь для энтерального питания) в связи с предполагаемым заболеванием. Проводилась метаболическая терапия: левокарнитин, цитофлавин, витамин В1 (по 300 мг/сут), убидекаренон. Получал также препараты железа (III) гидроксид полимальто-зат, глюконат кальция, гидрокарбонат натрия. В связи с развившимся острым бронхитом получал антибактериальную терапию — ампициллин/сульбактам.
09.03.2023 получены результаты секвенирования экзома: выявлен гомозиготный вариант нуклеотидной последовательности в гене PDНА1 (X-19359612-С-Е), приводящей к замене аминокислот 378-й позиции белка. Вариант описан в гомозиготной форме у пациентов с дефицитом пируватдегидрогеназы, в том числе как возникший de novo .
С 07.04.2023 ребёнок в стабильно тяжёлом состоянии без зависимости от кислорода переведён в отделение паллиативной медицинской помощи детям, где находился по конец апреля 2023 года. Согласно рекомендациям врача-диетолога, в питании ребёнка увеличена дотация белка энтерально до 2 г/кг в сутки.
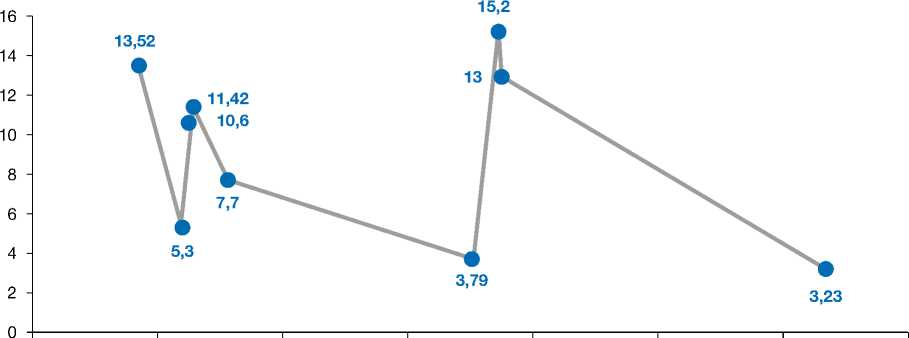
Осуществлялся постоянный динамический контроль метаболического состояния пациента с использованием кетометра и аммониметра (рис. 1, 2). В ходе обследования отмечались колебания уровня аммиака крови от 120 до 154 ммоль/л, на фоне лечения — снижение до 38 ммоль/л. Другие значимые показатели представлены в табл. 1.
Проведена телемедицинская консультация с медико-генетическим центром ФГАУ «Национальный медицинский исследовательский центр здоровья детей» Минздрава России (Москва). Диагноз и тактика ведения пациента согласованы.
Мальчик выписан домой на 37-й день пребывания в стационаре в удовлетворительном состоянии под диспансерное наблюдение участкового педиатра и врачей-узких специалистов с рекомендациями продолжить приём лечебной высокожировой низкоуглеводной сухой смеси для энтерального питания до 6–8 раз в сутки с коррекцией дозы смеси по результатам антропометрии. Назначена высокая доза витамина В1 (тиамина хлорид): по 10 таблеток (10 мг) 3 раза в день длительным курсом. Левокарнитин внутрь по 5 капель 3 раза в день постоянно. Убидекаренон внутрь по 6 капель 2 раза в день. Урсофальк суспензия по 250 мг/мл, 1 мл на ночь в течение месяца.
В возрасте 5 месяцев ребёнок поступил в плановом порядке в отделение паллиативной медицинской помощи детям СОКБ им. Середавина. Состояние тяжёлое по основному заболеванию.

7,4
7,35
7,3 7,28 7,29
7,37
7,38
7,3
7,31
7,38
7,25
m о
7,25
7,2
7,18
7,15
7,1
’ 7,16 7,09
7,05
День пребывания в стационаре • pH
Рис. 1. Показатель кислотно-щелочного состояния в период пребывания в стационаре. Норма кислотно-щелочного состояния 7,35–7,45.
Fig. 1. Indicator of acid-base condition — blood pH during hospitalization. Normal acid-base status 7.35–7.45.
05.12.22 24.01.23 15.03.23 04.05.23 23.06.23 12.08.23 01.10.23 20.11.23
День пребывания в стационаре лактат
Рис. 2. Показатели лактатдегидрогеназы за время наблюдения пациента.
Fig. 2. Lactate dehydrogenase indicators during the patient's follow-up from 17.01.2023 to June 10, 2023.
Неврологический статус при осмотре: голову не удерживает; отсутствие зрительного и слухового сосредоточения; отмечается непостоянная фиксация взгляда с периодически возникающим нистагмом и закатыванием глаз; мышечный тонус переменный с периодическим усилением дистонии. Проведён биохимический анализ крови (см. табл. 1).
После консультации врача-невролога (заведующая отделением для детей с поражением центральной нервной системы и нарушением психики Н.Н. Савельева) выставлен диагноз: «Нейродегене- ративное заболевание. Дефицит пируватдегидрогеназы Е1 альфа. Центральный тетрапарез, преимущественно правосторонний. Синдром инфантильных спазмов. Ротаторный нистагм». В лечение добавлен леветирацетам питьевой раствор 0,6 мл 3 раза в день (9:00, 14:00, 20:00), или 180 мл в сутки.
Согласно осмотру врача-диетолога (заведующая отделом лечебного питания Н.Н. Кузнецова), дополнительно к основному был выставлен диагноз белково-энергетической недостаточности лёгкой степени и задержки физического развития.
Таблица 1 / Table 1
Значимые показатели биохимического исследования крови / Significant indicators of biochemical blood testing
|
Результат |
Единица измерения |
Значение |
Норма/Патология |
Дата исследования |
|
Глюкоза |
ммоль/л |
3,4 |
Норма |
17.01.2023 |
|
Глюкоза |
ммоль/л |
4,09 |
Норма |
07.02.2023 |
|
Глюкоза |
ммоль/л |
5,8 |
Патология |
18.02.2023 |
|
Билирубин общий |
мкмоль/л |
9,8 |
Норма |
17.01.2023 |
|
Билирубин общий |
мкмоль/л |
9,1 |
Норма |
07.02.2023 |
|
Билирубин общий |
мкмоль/л |
5,8 |
Норма |
18.02.2023 |
|
С-реактивный белок |
мг/дл |
0 |
Норма |
17.01.2023 |
|
С-реактивный белок |
мг/дл |
0,9 |
Норма |
07.02.2023 |
|
С-реактивный белок |
мг/дл |
6,1 |
Патология |
18.02.2023 |
|
Аммоний |
мкмоль/л |
121,9 |
Патология |
17.01.2023 |
|
Аммоний |
мкмоль/л |
71,1 |
Патология |
07.02.2023 |
|
Лактатдегидрогеназа |
Ед/л |
169,8 |
Норма |
29.05.2023, 17:25 |
|
Натрий (Na) |
ммоль/л |
134,9 |
Патология |
29.05.2023, 17:25 |
|
Лактат |
ммоль/л |
3,79 |
Патология |
29.05.2023, 18:28 |
В отделении ребёнок продолжал получать питание высокожировой низкоуглеводной сухой смесью для энтерального питания (по 14,2 г смеси + 120 мл воды) с коррекцией дозы.
За период госпитализации проведено следующее медикаментозное лечение: парентерально : магния сульфат 25% 1,5 мл + р-р натрия хлорида 0,9% 100,0 мл, № 4; левокарнитин 10 мл + р-р натрия хлорида 0,9% 100,0 мл, № 5; цитофлавин 2,0 мл + р-р натрия хлорида 0,9% 50,0 мл, № 5; р-р тиамина хлорида 2,0 мл + р-р натрия хлорида 0,9% 10,0 мл 3 раза в день, 5 дней; внутрь : убидека-ренон по 6 капель 2 раза в день, масляный раствор колекальциферола по 2 капсулы в сутки, леветирацетам 100 мл по 0,6 мл 3 раза в день.
В июне 2023 года ребёнок в стабильном состоянии выписан домой под наблюдение участковых врачей (педиатра, невролога, диетолога, офтальмолога, ортопеда, врача паллиативной службы) с динамическим уровнем контроля лактата крови 1 раз в месяц, газов крови 1 раз в 2 недели, уровня кетонов в крови натощак через день, уровня бета-гидроксибутирата. Даны рекомендации по продолжению кетогенной диеты, приёму тиамина хлорида в виде порошков (тиамина хлорид 300 мг/сут), противосудорожной и метаболической терапии.
В возрасте 9 месяцев у мальчика отмечена незначительная положительная неврологическая динамика. Состояние стабильное, средней тяжести. Голову удерживает непостоянно, есть слабая реакция зрительного и слухового сосредоточения.
Нистагм отмечается периодически. Мышечная гипотония. Опора на ноги снижена.
Прогноз
Прогноз для жизни пациента на фоне проводимой терапии благоприятный.
ОБСУЖДЕНИЕ
Как правило, средний возраст постановки диагноза дефицита пируватдегидрогеназы и начала лечения составляет около 45 месяцев (средний возраст ~20 месяцев) [1, 21].
На сегодняшний день этиотропная терапия дефицита пируватдегидрогеназного комплекса не разработана, поэтому пациентам назначается патогенетическая и симптоматическая терапия. Одним из предлагаемых методов лечения является кетогенная диета, включающая большое количество жиров, благодаря которым организм начинает получать энергию за счёт разложения жирных кислот [22]. На фоне лечения снижается уровень лактата в крови, уменьшается частота судорог, нормализуется сон, потенцируются развитие речи, общей и мелкой моторики, снижается частота госпитализаций [21]. В некоторых исследованиях отмечается улучшение состояния при добавлении к проводимому лечению тиамина [23], однако в экспериментах не выявлено изменений суммарной ферментативной активности пируватдегидрогеназного комплекса при дефиците тиамина [24].
На фоне проводимой терапии отмечается значительная положительная динамика в нервно-психическом развитии ребёнка за период наблюдения. Следует отметить, что ранняя постановка диагноза и своевременно начатое лечение улучшают прогноз для пациентов с дефицитом пируватдегидрогеназы, что также отмечается в исследованиях других авторов [3, 21].
ЗАКЛЮЧЕНИЕ
Дефицит пируватдегидрогеназы — одна из форм тяжёлых наследственных митохондриальных болезней обмена, характеризующаяся нарушениями энергетического обмена и проявляющаяся широким спектром неврологических симптомов. Ранняя постановка диагноза имеет крайне важное значение для физического и нервно-психического развития детей с данной патологией, что обусловлено своевременным началом терапии. Несмотря на отсутствие высокоэффективного этиотропного лечения, в некоторых случаях, к которым относится и описываемый нами, улучшение клинического течения наблюдается при применении препаратов тиамина, а также соблюдении кетогенной диеты.
ДОПОЛНИТЕЛЬНАЯ ИНФОРМАЦИЯ
Источник финансирования. Авторы заявляют об отсутствия внешнего финансирования при подготовке статьи.
Список литературы Дефицит пируватдегидрогеназы у мальчика: клинический случай
- DeBrosse SD, Okajima K, Zhang S, et al. Spectrum of neurological and survival outcomes in pyruvate dehydrogenase complex (PDC) deficiency: Lack of correlation with genotype. Mol Genet Metab. 2012;107(3):394–402. doi: 10.1016/j.ymgme.2012.09.001
- Horga A, Woodward CE, Mills A, et al. Differential phenotypic expression of a novel PDHA1 mutation in a female monozygotic twin pair. Hum Genet. 2019;138(11-12):1313–1322. EDN: NTSHMV doi: 10.1007/s00439-019-02075-9
- Blass JP, Avigan J, Uhlendorf BV. A defect in pyruvate decarboxylase in a child with an intermittent movement disorder. J Clin Invest. 1970;49(3):423–432. doi: 10.1172/JCI106251
- Imbard A, Butron A, Veco S, et al. Molecular characterization of 82 patients with pyruvate dehydrogenase complex deficiency. Structural consequences of new amino acid substitutions in the E1 protein. Mol Genet Metab. 2011;104(4):507–516. doi: 10.1016/j.ymgme.2011.08.008
- Adeva M, González-Lucán M, Seco M, Donapetry C. Enzymes involved in l-lactate metabolism in humans. Mitochondrion. 2013;13(6):615–629. EDN: YDORFL doi: 10.1016/j.mito.2013.08.011
- Brown GK, Otero LJ, LeGris M, Brown RM. Pyruvate dehydrogenase deficiency. J Med Genet. 1994;31(11):875–879. doi: 10.1136/jmg.31.11.875
- Kubiak GM, Tomasik AR, Bartus K, et al. Lactate in cardiogenic shock: Current understanding and clinical implications. J Physiol Pharmacol. 2018;69(1):15–21. doi: 10.26402/jpp.2018.1.02
- Hayashida K, Suzuki M, Yonemoto N, et al.; SOS-KANTO 2012 Study Group. Early lactate clearance is associated with improved outcomes in patients with postcardiac arrest syndrome: A prospective, multicenter observational study (SOS-KANTO 2012 Study). Crit Care Med. 2017;45(6): e559–e566. doi: 10.1097/CCM.0000000000002307
- Li X, Yang Y, Zhang B, et al. Lactate metabolism in human health and disease. Signal Transduct Target Ther. 2022;7(1):305. EDN: VGHXFC doi: 10.1038/s41392-022-01151-3
- Vilarino L, Nogueira S. PCR in the analysis of clinical samples: Prenatal and postnatal diagnosis of congenital metabolic disorders. Methods of Mol Biol. 2017;(1620):213–224. doi: 10.1007/978-1-4939-7060-5_15
- Pavlu-Pereira H, Silva MJ, Florindo C, et al. Pyruvate dehydrogenase complex deficiency: Updating the clinical, metabolic and mutational landscapes in a cohort of Portuguese patients. Orphanet J Rare Dis. 2020;15(1):298. EDN: RLSIBL doi: 10.1186/s13023-020-01586-3
- Sperl V, Fleuren R, Freisinger P., et al. The spectrum of pyruvate oxidation defects in the diagnosis of mitochondrial disorders. J Inherit Metab Dis. 2015;38(3):391–403. doi: 10.1007/s10545-014-9787-3
- Bedoyan JK, Hecht L, Zhang S, et al. A novel null mutation in the pyruvate dehydrogenase phosphatase catalytic subunit gene (PDP1) causing pyruvate dehydrogenase complex deficiency. JIMD Rep. 2019;48(1):26–35. doi: 10.1002/jmd2.12054
- Maj MC, Cameron JM, Robinson BH. Pyruvate dehydrogenase phosphatase deficiency: Orphan disease or an under-diagnosed condition? Mol Cell Endocrinol. 2006;249(1-2):1–9. doi: 10.1016/j.mce
- Gupta N, Rutledge C. Pyruvate dehydrogenase complex deficiency: An unusual cause of recurrent lactic acidosis in a paediatric critical care unit. J Crit Care Med (Targu Mures). 2019;5(2):71–75. doi: 10.2478/jccm-2019-0012
- Giribaldi G, Doria-Lamba L, Biancheri R, et al. Intermittent-relapsing pyruvate dehydrogenase complex deficiency: A case with clinical, biochemical, and neuroradiological reversibility. Dev Med Child Neurol. 2012;54(5):472–476. doi: 10.1111/j.1469-8749.2011.04151.x
- Barnerias C, Saudubray JM, Touati G, et al. Pyruvate dehydrogenase complex deficiency: four neurological phenotypes with differing pathogenesis. Dev Med Child Neurol. 2010;52(2):1–9. doi: 10.1111/j.1469-8749.2009.03541.x
- Egloff C, Eldin de Pecoulas A, Mechler C, et al. Prenatal sonographic description of fetuses affected by pyruvate dehydrogenase or pyruvate carboxylase deficiency. Prenat Diagn. 2018. doi: 10.1002/pd.5282
- Meldau S, Fratter C, Bhengu LN, et al. Pitfalls of relying on genetic testing only to diagnose inherited metabolic disorders in non-western populations: 5 Cases of pyruvate dehydrogenase deficiency from South Africa. Mol Genet Metab Rep. 2020;(24): 100629. doi: 10.1016/j.ymgmr.2020.100629
- Karissa P, Simpson T, Dawson SP, et al. Comparison between dichloroacetate and phenylbutyrate treatment for pyruvate dehydrogenase deficiency. Br J Biomed Sci. 2022;(79):10382. EDN: TYRFOF doi: 10.3389/bjbs.2022.10382
- Sofou K, Dahlin M, Hallböök T, et al. Ketogenic diet in pyruvate dehydrogenase complex deficiency: Short- and long-term outcomes. J Inherit Metab Dis. 2017;40(2):237–245. doi: 10.1007/s10545-016-0011-5
- Scholl-Bürgi S, Höller A, Pichler K, et al. Ketogenic diets in patients with inherited metabolic disorders. J Inherit Metab Dis. 2015;38(4):765–773. doi: 10.1007/s10545-015-9872-2
- Ogawa E, Hishiki T, Hayakawa N, et al. Ketogenic diet in action: Metabolic profiling of pyruvate dehydrogenase deficiency. Mol Genet Metab Rep. 2023;(35):100968. EDN: NTRUWY doi: 10.1016/j.ymgmr.2023.100968
- Elnageh KM, Gaitonde MK. Effect of a deficiency of thiamine on brain pyruvate dehydrogenase: Enzyme assay by three different methods. J Neurochem. 1988;51(5):1482–1489. doi: 10.1111/j.1471-4159.1988.tb01115.x
