Diagnostic Challenges in Hereditary Angioedema with Normal C1-Inhibitor Levels: A Clinical Case Report

Author: Volf A., Tabynbayeva D., Nagornaya A., Kerimbay S., Nalobina M., Dedova O., Koshkarbayeva B., Knaus A., Izmailovich M.
Journal: Juvenis scientia @jscientia
Section: Клинические случаи
Article in issue: 5 т.11, 2025.
Free access
Hereditary angioedema with normal C1‑inhibitor (HAE-nC1INH) remains a diagnostic challenge due to normal complement parameters and clinical similarity to histamine-mediated angioedema, frequently leading to misdiagnosis and delayed initiation of appropriate therapy. The aim of this report was to demonstrate diagnostic reasoning in a clinically suspected case of HAE-nC1INH. We present a clinical case of a 25‑year-old woman with recurrent, non-urticarial edema involving the face and extremities, resistant to antihistamines and systemic corticosteroids. Clinical presentation, family history, and laboratory findings were analyzed, including complement C4 level, C1‑inhibitor activity, thyroid hormone levels, and serum 25‑hydroxyvitamin D. The patient exhibited normal C4 levels and borderline C1‑inhibitor activity in combination with a typical clinical phenotype and positive family history, supporting a probable diagnosis of HAE-nC1INH. A pronounced vitamin D deficiency was also identified. An allergic mechanism of angioedema was considered unlikely. This case underscores the importance of heightened clinical awareness in patients with recurrent angioedema of unclear origin. Early recognition of HAE-nC1INH facilitates timely initiation of targeted therapy and prevents prolonged ineffective treatment.
Hereditary angioedema, C1 esterase inhibitor, diagnosis, clinical case, facial edema
Short address: https://sciup.org/14134411
IDR: 14134411 | DOI: 10.32415/jscientia_2026_11_5_27-34
Диагностические трудности при наследственном ангиоотёке с нормальным уровнем С1-ингибитора: клинический случай
Наследственный ангиоотёк с нормальным уровнем С1‑ингибитора (НАО-nC1INH) относится к редким формам ангиоотёка и характеризуется значительными диагностическими трудностями. Отсутствие специфических лабораторных маркеров и клиническое сходство с гистамин-опосредованными отёками нередко приводят к ошибочной диагностике, задержке установления правильного диагноза и назначению неэффективной терапии. В работе представлен клинический случай 25‑летней пациентки с многолетними рецидивирующими эпизодами отёка лица и конечностей, не отвечающими на терапию антигистаминными препаратами и глюкокортикостероидами. Проведён комплексный анализ клинических проявлений, семейного анамнеза и лабораторных данных, включая уровень компонента комплемента C4, активность С1‑ингибитора, показатели функции щитовидной железы и концентрацию 25‑гидроксивитамина D. У пациентки выявлены нормальные показатели C4 и пограничная активность С1‑ингибитора на фоне характерной клинической картины и отягощённого семейного анамнеза, что позволило заподозрить НАО-nC1INH. Дополнительно установлен выраженный дефицит витамина D. Аллергическая природа отёков была признана маловероятной. Представленный клинический случай подчёркивает необходимость высокой клинической насторожённости при оценке рецидивирующих отёков. Своевременная диагностика НАО-nC1INH позволяет перейти к патогенетически обоснованной терапии и избежать длительного неэффективного лечения.
Text of the scientific article Diagnostic Challenges in Hereditary Angioedema with Normal C1-Inhibitor Levels: A Clinical Case Report
Hereditary angioedema (HAE) is a rare autosomal dominant disorder caused by deficiency or dysfunction of C1 esterase inhibitor (C1-INH), a protease that regulates complement, contact, fibrinolytic, and coagulation systems [1]. The global prevalence is estimated at approximately 1 in 50,000 individuals, although many cases remain unrecognized due to atypical manifestations and frequent misdiagnosis as allergic angioedema [2].
HAE is classified into three main types based on C1-INH concentration and function. Type I (approximately 85% of cases) is characterized by reduced levels of C1-INH, whereas Type II (around 15%) involves normal but functionally inactive protein [3, 4]. Both forms are bradykinin-mediated, presenting as recurrent, non-pitting edema of the skin, gastrointestinal tract, or airways [5].
The third form, HAE with normal C1-INH (HAE-nC1INH), occurs despite normal quantitative and functional levels of the inhibitor [6]. It predominantly affects women and may be estrogen-dependent. Identified gene mutations include F12, PLG, and AN-GPT1, which lead to excessive bradykinin generation, vascular permeability, and tissue edema [7].
Timely recognition of HAE remains challenging, particularly in cases with normal complement levels, where symptoms mimic histamine-mediated angio-edema. This report illustrates the clinical reasoning and diagnostic process that led to the suspicion of HAE-nC1INH during medical training.
The main types of bradykinin-mediated angioede-ma, their mechanisms, and clinical characteristics are summarized in Table 1.
CLINICAL CASE
A 25-year-old female student presented with recurrent episodes of swelling affecting the face and extremities, often following physical exertion or upper respiratory infections. Episodes were accompanied by
Table 1
Classification and key characteristics of bradykinin-mediated angioedema [1]
|
Type of angioedema |
Pathophysiological mechanism |
Main genetic / biochemical causes |
Key clinical features |
|
Hereditary angioedema type I |
Deficiency of C1 esterase inhibitor → uncontrolled activation of the kallikrein-kinin cascade → bradykinin overproduction |
SERPING1 mutations → low C1-INH level |
Recurrent edema of skin, gastrointestinal tract, or airways; positive family history; onset in childhood or adolescence |
|
Hereditary angioedema type II |
Dysfunction of C1 esterase inhibitor despite normal protein level |
SERPING1 mutations → functionally inactive C1-INH |
Similar symptoms to HAE I; laboratory finding: normal level, low activity of C1-INH |
|
HAE with normal C1-INH |
Increased activation of factor XII and plasma kallikrein with excessive bradykinin formation despite normal C1-INH |
Mutations in F12 , PLG , ANGPT1 , KNG1 , MYOF , HS3ST6 |
Predominantly in females; may be estrogen-dependent; recurrent edema of face, lips, tongue, and abdomen |
|
Acquired angioedema |
Secondary consumption or autoimmune inactivation of C1-INH |
Lymphoproliferative or autoimmune disorders; presence of anti-C1-INH antibodies |
Late onset ( > 40 years); absence of family history; recurrent non-pruritic swelling |
|
Drug-induced bradykinin-mediated angioedema |
Impaired degradation of bradykinin due to inhibition of kininase II |
ACE inhibitors, DPP-IV inhibitors |
Isolated facial or laryngeal edema, often in elderly patients, without urticaria or itching |
Table 2
Laboratory parameters of the patient with suspected hereditary angioedema
|
Parameter |
Result |
Reference range |
|
C4 complement component |
0.26 g/L |
0.15–0.57 |
|
C1 esterase inhibitor activity |
1.26 U/mL |
0.7–1.3 |
|
Free T3 |
4.94 pmol/L |
1.8–4.2 |
|
Free T4 |
0.816 pmol/L |
0.89–1.76 |
|
25-hydroxyvitamin D (25(OH)D) |
5.1 ng/mL |
> 10 |
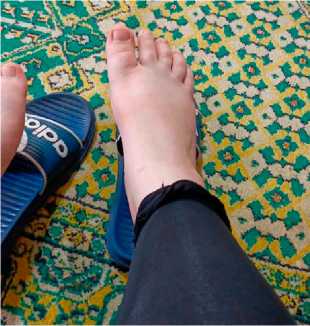
Fig. 1. Recurrent edema of the right hand and foot in a patient with hereditary angioedema
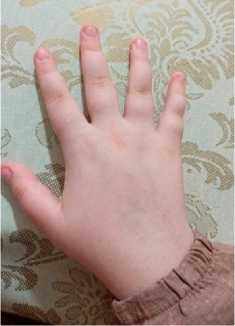
throat tightness and transient dyspnea. Typical manifestations of swelling are shown in Figure 1.
The patient had a history of atopic dermatitis since childhood. Edema episodes had been occurring for the past 10 years, with a frequency of one to two episodes per year. These episodes were unresponsive to antihistamines or corticosteroids and resolved spontaneously within several days. The most recent episode developed after acetaminophen intake.
Family history : the patient’s father and sister experienced episodes of seasonal rhinitis and mild, selflimiting facial swelling.
Circumstances of presentation : during a practical allergology class, the instructor observed marked facial swelling and somnolence in the student, who was under antihistamine therapy at that time. She was referred for additional examination at the university clinical base.
Status praesens: the general condition was satisfac- tory. Skin examination revealed mild erythema, dryness, and thickened areas with excoriations on the flexural surfaces and perioral region. Positive pink dermographism was noted. The tongue was coated with a yellowish deposit. No urticaria or pruritus was observed. Vital signs, urination, and bowel habits were normal. The results of laboratory investigations are summarized in Table 2.
The results show borderline C1-INH activity with normal C4 levels and a pronounced (25(OH)D) deficiency, supporting the need for genetic verification of HAE in clinically suspected cases.
DIAGNOSIS
Primary: (T78.3) Angioneurotic edema, probable hereditary (confirmation pending).
Comorbidities: (L20.8) Atopic dermatitis; (E55.9) Vitamin D deficiency; (E05.90) Thyroid dysfunction (by laboratory data).
DISCUSSION
The presented case highlights the diagnostic complexity of HAE-nC1INH. Unlike classical forms, laboratory parameters such as C4 and C1-INH activity may remain within normal limits, delaying recognition and misdirecting therapy toward allergic causes. This diagnostic ambiguity is further compounded by overlapping symptoms with histamine-mediated an-gioedema, which often results in prolonged use of ineffective treatments. Indeed, in large reviews, HAE-nC1INH is noted for its heterogeneity in presentation, overlap with HAE type I/II, and a lack of well-defined biomarkers, which often lead to diagnostic delays of 5–10 years or more [8, 9].
In international consensus, HAE-nC1INH is increasingly recognized as a distinct entity requiring tailored diagnostic pathways. Genetic variants in F12, PLG, AN-GPT1, KNG1, HS3ST6, MYOF, and others are implicated in pathogenesis via dysregulation of the kallikrein-bradykinin axis [10]. Recent whole-exome sequencing studies have expanded this spectrum by identifying novel MYOF and HS3ST6 variants correlated with more protracted and refractory edema episodes [11]. These findings reinforce the view that negative genetic testing does not exclude HAE-nC1INH but argues for continued investigation in phenotypically consistent cases.
Figure 2 illustrates the pathophysiologic cascade leading from factor XII activation to bradykinin release and increased vascular permeability [12].
Therapeutically, management of HAE-nC1INH largely follows guidelines extrapolated from HAE type I/II, given the limited trial data specific to the normal C1-INH subgroup [13, 14]. Agents targeting the bradykinin pathway — such as C1-INH replacement, bradykinin B2 receptor antagonists, and plasma kal-likrein inhibitors — are considered. Novel therapies are emerging: garadacimab, a monoclonal antibody to factor XIIa, showed favorable safety and efficacy signals in a small HAE-nC1INH cohort [15]. The CASPIAN study of lanadelumab in non-histaminergic nC1INH angioedema patients demonstrated safety consistent with prior HAE trials and a trend toward attack reduction, although statistical significance was not achieved [16]. A recent narrative review outlines additional advances in prophylactic and on-demand treatments,
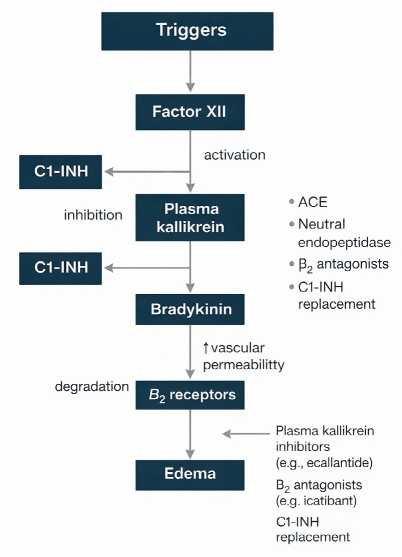
Fig. 2. Mechanisms of hereditary angioedema (according to Lima H. et al. [12]).
projecting a shift toward personalized regimens [17].
Our case underscores the need for heightened clinical suspicion in recurrent, nonpruritic, non-urti-carial edema, particularly in young women triggered by exercise, hormonal fluctuations, or minor insults. A stepwise diagnostic approach — ruling out hista-minergic etiologies, assessing complement assays, and proceeding to targeted genetic panels — is advocated [18]. Early identification allows initiation of appropriate therapy and avoids ineffective empirical antihistamine or corticosteroid regimens.
Finally, the psychological burden and quality-of-life impairment in HAE patients is increasingly recognized. Recent narrative reviews report elevated rates of anxiety, depression, and sleep disturbance in HAE populations, highlighting the need for holistic care beyond attack control [19]. In such rare cases as ours, multidisciplinary follow-up — with immunology, genetics, dermatology, and psychology input — optimizes patient outcomes.
CONCLUSION
This case demonstrates the need for heightened clinical vigilance when assessing recurrent edema of unknown origin. Early diagnosis of hereditary an-gioedema depends on thorough history taking, family evaluation, and directed laboratory and genetic testing. Incorporating such cases into the medical education process enhances the diagnostic competence of healthcare professionals and awareness of rare immunologic diseases.
Funding: The authors declare that no funding was received.
Conflict of Interest: The authors declare no conflict of interest.
Ethics Statement: Written informed consent was obtained from the patient for the publication of this clinical case and any accompanying images. The patient’s anonymity and confidentiality were fully preserved in accordance with ethical standards.
Author Contributions: Conceptualization, M. I.; methodology, O. D. and B. K.; validation, A. K. and A. V.; investigation, D. T., A. N., S. K., and M. N.; resources, B. K.; data curation, O. D.; writing - original draft preparation, M. I., O. D., A. N., and A. V.; writing - review and editing, M. I. and A. V.; visualization, M. I.; supervision, B. K.; project administration, M. I.; funding acquisition – not applicable. All authors were equally involved in the writing of this article.
