Дизайн, молекулярный докинг и анализ «структура–свойство» новых производных комбретастатина А-4

Автор: Семенов А.В., Балакирева О.И., Агальцова А.С., Афанасьев С.М.
Журнал: Огарёв-online @ogarev-online
Рубрика: Технические науки
Статья в выпуске: 1 т.14, 2026 года.
Бесплатный доступ
Введение. Одним из наиболее перспективных направлений таргетного подхода к лечению злокачественных новообразований является ингибирование полимеризации тубулина. В связи с этим, целью настоящего исследования является молекулярный дизайн новых производных комбретастатина А-4 для выявления соединений с высокой аффинностью к колхициновому сайту β-тубулина. Материалы и методы. Молекулярный докинг выполнялся в программном комплексе Glide (модуль Schrödinger Maestro). В качестве рецептора использована кристаллографическая структура комплекса тубулина с комбретастатином A4 (PDB ID 5LYJ). Для выявления зависимостей «структура–свойство» были вычислены следующие дескрипторы: количество гидроксильных и метоксильных групп; наличие C=C-связей, циклопропановых и 1,2,5-оксадиазольных колец. ADME характеристики были рассчитаны с использованием ресурса ADMETlab. Результаты исследования. Из 96 рассмотренных соединений 7 молекул продемонстрировали лучшие (более отрицательные) значения GlideScore, чем комбретастатин. Наличие метоксильных групп отрицательно коррелирует со значением GlideScore, внося основной вклад в аффинность за счет гидрофобных взаимодействий с сайтом связывания. Гидроксильные группы также способствует улучшению связывания с мишенью за счет формирования дополнительных водородных связей. Замена двойной связи на циклопропановый фрагмент приводит к получению соединений с сопоставимой аффинностью (GlideScore ≈ – 8,2 ккал/моль). Напротив, оксадиазольные аналоги в среднем демонстрируют худшие показатели связывания. Обсуждение и заключение. Соединения, содержащие цис-алкеновый мостик и комбинацию гидрокси- и метокси-заместителей, продемонстрировали значения GlideScore, превосходящие комбретастатин A-4, что делает их кандидатами для дальнейшего синтеза и экспериментального изучения in vitro.
Комбретастатин А-4, аналоги, молекулярный докинг, ингибиторы полимеризации тубулина, противоопухолевые свойства
Короткий адрес: https://sciup.org/147253576
IDR: 147253576 | УДК: 547.562 | DOI: 10.15507/2311-2468.014.202601.064-073
Design, Molecular Docking, and Structure–Property Analysis of Novel Combretastatin A-4 Derivatives
Introduction. One of the most promising areas of a targeted approach to the treatment of malignant neoplasms is the inhibition of tubulin polymerization. In this regard, the aim of this study is the molecular design of new derivatives of combretastatin A-4 to identify compounds with high affinity for the colchicine site of β-tubulin. Materials and methods. Molecular docking was performed in the Glide software package (Schrödinger Maestro module). The crystallographic structure of the tubulin-combretastatin A4 complex (PDB ID 5LYJ) was used as a receptor. To identify the structure–property relationships, the following descriptors were calculated: the number of hydroxyl and methoxyl groups; the presence of C=C bonds, cyclopropane and 1,2,5-oxadiazole rings. The ADME characteristics were calculated using the ADMETlab resource. Results. Of the 96 compounds examined, 7 molecules demonstrated better (more negative) GlideScore values than combretastatin. The presence of methoxyl groups negatively correlates with the value of GlideScore, making the main contribution to affinity due to hydrophobic interactions with the binding site. Hydroxyl groups also contribute to improved binding to the target due to the formation of additional hydrogen bonds. Replacing the double bond with a cyclopropane fragment results in compounds with comparable affinity (GlideScore ≈ – 8.2 kcal/mol). In contrast, oxadiazole analogues, on average, exhibit worse binding rates. Discussion and conclusion. Compounds containing a cis-alkene bridge and a combination of hydroxy and methoxy substituents demonstrated GlideScore values superior to combretastatin A-4, which makes them candidates for further synthesis and experimental study in vitro.
Текст научной статьи Дизайн, молекулярный докинг и анализ «структура–свойство» новых производных комбретастатина А-4
eISSN 2311-2468
EDN:
Злокачественные новообразования остаются одной из ведущих причин смертности во всем мире. Это обусловлено их фундаментальным свойством – неконтролируемой пролиферацией и нарушением регуляции клеточного цикла. Современная стратегия тар-гетной терапии, направленной на специфические молекулярные структуры опухолевых клеток, позволяет достичь избирательного подавления роста новообразований при снижении системной токсичности. Одним из наиболее перспективных направлений таргет-ного подхода является ингибирование полимеризации тубулина – структурного белка, формирующего микротрубочки, которые обеспечивают поддержание клеточной формы, внутриклеточный транспорт и процесс митоза [1].
Нарушение динамической нестабильности микротрубочек приводит к остановке клеточного цикла и индукции апоптоза, что подтверждает тубулин как высоковалидную мишень для противоопухолевых препаратов [2]. К числу классических ингибиторов микротрубочек относятся паклитаксел, винкристин и колхицин, связывающиеся с различными сайтами β -тубулина (таксановым, винка-алкалоидным и колхициновым соответственно). Особый интерес вызывают соединения, взаимодействующие с колхициновым сайтом, поскольку они способны преодолевать множественную лекарственную устойчивость [3].
Целью работы является выявление соединений с высокой аффинностью к колхициновому сайту β -тубулина, представляющих интерес для дальнейшего синтеза и биологической оценки.
ОБЗОР ЛИТЕРАТУРЫ
Одним из наиболее изученных ингибиторов этого типа выступает комбретаста-тин A-4 (CA-4) (1) – природный стильбеноид, выделенный из Combretum caffrum 1. Связываясь с колхициновым сайтом β -тубулина, соединение (1) индуцирует деполимеризацию микротрубочек, проявляя выраженную антимитотическую и антиваскулярную активность [4]. Простая структура и высокая цитотоксичность обусловили использование (1) как ведущего фармакофора для разработки новых антимитотических соединений. Исследования в области «структура–активность» ( Specific Absorption Rate , SAR ) показали, что для реализации активности критически важны: 3,4,5-триметоксифенильный фрагмент (кольцо A), цис -конфигурация ароматических колец относительно этиленового моста и 3-гидрокси-4-метокси-заместитель в фенильном кольце B [5].
Однако клиническое применение (1) ограничено его низкой биодоступностью, вызванной изомеризацией активной цис -формы в термодинамически стабильную, но малоактивную транс -форму in vivo . Для устранения данного недостатка предложены структурные модификации, включающие замену этиленового моста изостерными циклическими или гетероциклическими фрагментами (триазольными [6], оксазольными [7], изоксазольными [8] и др.), что предотвращает изомеризацию и повышает противоопухолевую активность.
66 Технические науки
Важным инструментом рационального дизайна аналогов CA-4 является молекулярный докинг, позволяющий моделировать пространственную ориентацию лигандов в активном сайте белка, оценивать энергию связывания и выявлять ключевые межмолекулярные взаимодействия.
МАТЕРИАЛЫ И МЕТОДЫ
Молекулярное моделирование выполнено для серии аналогов комбретастатина, в которых варьировались: количество и положение гидроксильных и метоксильных заместителей на бензольных или пиридиновых кольцах; природа центрального фрагмента, соединяющего арильные кольца – цис -алкеновая связь, циклопропановое или оксадиа-зольное кольцо.
Исследование направлено на дизайн и молекулярный докинг новых производных CA-4, основанных на стильбазольных структурах — аналогах цис -ресвератрола (2), где этиленовый мост заменён изостерными циклическими фрагментами (3,4) (рисунок 1).
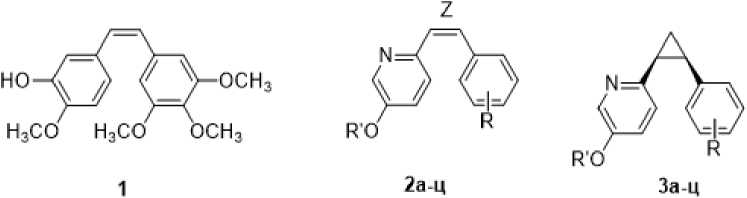
R'=H
R=H (a)
R=2-0H (6)
R=3-0H (в)
R=4-0H (r)
R=2.3- R=2,4-(OH)2 (e) R=2.5-(OH)2(ж) R=3.4- R=3.5- R=2.3,4-(OH)3 (к) R=3.4,5-(OH)3 (л) R'=CH3: R=H (m) R=2-OCH3(h) R=3-OCH3 (o) R=4-OCH3(n) R=2.3-(OCH3)2 (p) R=2.4-(OCH3)2(c) R=2.5-(OCH3)2(t) R=3s4-(OCH3)2 (y) R=3.5-(OCH3)2 (ф) R=2,3.4-(OCH3)3 (x) R=3,4,5-(OCH3)3 (Ц) 4а-ц Р и с . 1 . Структурные формулы исследуемых соединений F i g . 1 . Structural formulas of the researched compounds Источник: материалы рисунков и таблиц подготовлены авторами по итогам исследования Source: the materials of the figures and tables were prepared by the authors based on the results of the study Все структуры были подготовлены в формате SMILES и конвертированы в трехмерные модели с использованием пакета Schrödinger Suite 2023-4. Расчеты проводились на рабочей станции под управлением операционной системы Windows 11 Pro версии 25Н2 с процессором AMD Ryzen 7 PRO 8845HS, объемом оперативной памяти 16 Гб и графическим ускорителем Radeon 780M Graphics. Геометрия соединений оптимизировалась в модуле LigPrep с учетом протонирования при pH 7,0 ± 0,5 и стандартных параметров силового поля OPLS4. В процессе подготовки генерировались все возможные стереоизомеры для хиральных центров, стереохимия которых не была явно задана во входных структурах, а также учитывалось образование возможных таутомеров. Молекулярный докинг выполнялся в программном комплексе Glide2 (модуль Schrödinger Maestro) [9]. В качестве рецептора использована кристаллографическая структура комплекса тубулина с комбрета-статином A-4 (PDB ID 5LYJ) [10]. Белковая структура была подготовлена с помощью Protein Preparation Wizard (реализовались оптимизация цепей, удаление воды, добавление отсутствующих атомов боковых цепей, минимизация при OPLS4). Минимизация проводилась с критерием сходимости по среднеквадратичному отклонению (Root– Mean–Square Deviation, RMSD) 0,30 Å. Сетка докинга центрировалась по положению исходного лиганда (1) с размером 20×20×20 Å, включающим весь гидрофобный карман β-тубулина. Докинг осуществлялся в режиме жесткого рецептора. Режимом для докинга был выбран Standard Precision (SP). Для каждой молекулы сохранялось по пять лучших конформеров, ранжированных по суммарному значению GlideScore и DockingScore. В качестве эталонного значения использовались результаты для комбретастатина (1) из того же расчета: GlideScore = - 8,888. Для выявления зависимостей «структура–свойство» были вычислены простые дескрипторы: – количество гидроксильных и метоксильных групп; – наличие C=C-связей между sp²-центрами; – наличие циклопропановых фрагментов; – наличие 1,2,5-оксадиазольных колец. Статистический анализ проводился с использованием Python (библиотеки Pandas, Scikit-learn, Matplotlib). Корреляционный анализ реализовывался с расчетом коэффициента корреляции Пирсона. Полученные p-значения корректировались для учета множественного сравнения с помощью метода Бенджамини-Хохберга (False Discovery Rate, FDR). Перед проведением анализа главных компонентов (Principle Component Analysis, PCA) исходные данные были стандартизированы путем вычитания среднего и масштабирования до единичной дисперсии (StandardScaler). Первые два главных компонента объясняют 81 % (PC1 = 58 %, PC2 = 23 %) общей дисперсии данных. ADME характеристики рассчитывались с использованием ресурса ADMETlab [11]. РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ В проанализированной серии присутствовали соединения с варьирующимся числом кислородсодержащих заместителей (от 1 до 5). Значения GlideScore находились в диапазоне от - 9,68 до – 8,26 ккал/моль, что сопоставимо и в ряде случаев превосходит результаты для эталонного комбретастатина (- 8,888 ккал/моль). Из 96 рассмотренных соединений 7 молекул продемонстрировали лучшие (более отрицательные) значения GlideScore, чем комбретастатин (табл.1). Это указывает на сохранение и возможное улучшение ключевых взаимодействий в активном сайте при определенных модификациях. 68 Технические науки Таблица 1. Результаты расчетов Table 1. Calculation results Соединение / Compound GlideScore, ккал/моль / GlideScore, kcal/mol M, г/моль / M, g/mol Log P o/w Log S TPSA, Å2 2ж – 9,675 229,07 2,03 – 2,43 73,58 2и – 9,393 229,07 2,17 – 3,05 73,58 3д – 9,103 243,09 1,93 – 2,84 73,58 2к – 9,027 245,07 1,71 – 2,61 93,81 2в – 8,981 213,08 2,66 – 3,36 53,35 3к – 8,954 259,08 1,62 – 2,08 93,81 3б – 8,939 227,09 2,30 – 2,76 53,35 2е – 8,866 229,07 2,13 – 2,69 73,58 2д – 8,822 229,07 2,00 – 2,71 73,58 4в – 8,820 255,06 2,54 – 3,56 92,27 1 - 8,888 316,35 2,87 – 3,83 57,15 Кроме того, для указанных соединений были рассчитаны основные ADME характеристики. Большинство соединений, представленных в таблице 1, имеют умеренную липофильность, среднюю растворимость и среднюю площадь полярной поверхности, что соответствует приемлемым ADME характеристикам для удовлетворительной пероральной биодоступности. На рисунке 2 представлено расположение в сайте связывания соединений каждого ряда, показавших лучшую аффинность. Р и с . 2 . Связывание соединений в колхициновом сайте β-тубулина: I – наложение структур 1 (красный), 2ж (оранжевый), 3д (зеленый), 4в (желтый); II – взаимодействия структур в сайте связывания F i g . 2 . Binding of compounds at the colchicine site of β-tubulin: I – superposition of structures 1 (red), 2ж (orange), 3д (green), 4в (yellow); II – interactions of structures at the binding site Анализ взаимодействия комбретастатина A-4 и его аналогов с тубулином показал, что все соединения связываются в колхициновом сайте и сохраняют ключевые фармакофорные взаимодействия. При этом для рассматриваемых производных фиксируются дополнительные контакты в форме водородных связей, что может обеспечивать повы- шенную аффинность связывания. Таким образом, аналоги являются эффективными био-изостерными заменами исходного соединения, демонстрируя перспективный профиль взаимодействий для дальнейшей структурной оптимизации в качестве ингибиторов полимеризации тубулина. Корреляционный анализ выявил взаимосвязь между структурными особенностями молекул и энергией связывания. Полученные результаты согласуются с литературными данными, подчеркивающими критическую важность метокси-групп, особенно в 3,4,5-положениях кольца А, для взаимодействия с колхициновым сайтом. Гидроксильные группы также показали умеренную положительную корреляцию, что говорит об их способности формировать дополнительные водородные связи, однако их избыток может увеличивать полярность молекулы, потенциально ухудшая связывание с преимущественно гидрофобным карманом. Замена цис-алкенового моста на циклопропановое кольцо не ухудшала энергию связывания. Одно из лучших значений GlideScore (- 9,10 ккал/моль) наблюдалось именно для циклопропанового аналога с двумя гидроксильными группами в орто- и мета-положениях. Это подтверждает возможность использования циклопропана как пространственного биоизостера цис-двойной связи, сохраняющего оптимальную ориентацию арильных колец. Данный подход активно используется для стабилизации биоактивной конформации и улучшения фармакокинетических свойств. В то же время замена на оксадиазольное звено ухудшала показатели, вероятно, вследствие нарушения планарности системы и дестабилизации взаимодействий с аминокислотными остатками в активном кармане. Тем не менее, некоторые исследования показывают, что правильная функционализация гетероциклических линкеров может компенсировать этот эффект и привести к высокоактивным соединениям. Наилучшее соединение в серии (2ж) показало GlideScore = - 9,675 ккал/моль, что на 0,787 ккал/моль лучше эталонного комбретастатина. Эта структура сохраняет цис-конфигурацию и содержит по одной гидроксильной группе на каждом арильном кольце, что обеспечивает симметричное взаимодействие с белком. Таким образом, минимальная модификация, не нарушающая геометрию и распределение доноров/акцепторов, может улучшить связывание. Иерархическая кластеризация (Ward linkage) выделила три устойчивых кластера, различающиеся как по типу мостика, так и по числу донорных групп. Средние значения GlideScore для этих кластеров представлены в таблице 2. Таблица 2. Средние значения GlideScore по типу мостика, ккал/моль Table 2. Average GlideScore values by bridge type, kcal/mol Тип мостика / Bridge type Среднее ± SD / Mean ± SD Медиана / Median Характерные признаки / Characteristic features Цис-C=C / Cis-C=C – 8,54 ± 0,27 – 8,53 Сохранённое π-сопряжение / Saved π-conjugation Циклопропан / Cyclopropane – 8,21 ± 0,33 – 8,20 Умеренно жёсткая цис-геометрия, потеря сопряжения / Moderately rigid cis geometry, loss of coupling Оксадиазол / Oxadiazole – 7,98 ± 0,31 – 8,00 Гетероциклический изостер, более выраженное искажение геометрии / Heterocyclic isostere, more pronounced distortion of geometry Анализ показал, что сохранение цис-C=C-моста обеспечивает наилучшее пространственное связывание и наивысшую аффинность. Циклопропановый мостик является приемлемой заменой, сохраняя цис-ориентацию с умеренной потерей эффективности связывания. Оксадиазольные производные образуют отдельный кластер с наихудшими показателями, что указывает на необходимость дальнейшей оптимизации их структуры. ОБСУЖДЕНИЕ И ЗАКЛЮЧЕНИЕ Комплексный анализ методом молекулярного докинга серии новых аналогов комбре-тастатина A-4 позволил выявить ключевые зависимости «структура–аффинность»: Наличие метоксильных групп отрицательно коррелирует со значением GlideScore, внося основной вклад в аффинность за счет гидрофобных взаимодействий с сайтом связывания. Гидроксильные группы также демонстрируют благоприятную, хотя и более слабую, отрицательную корреляцию, выступая в роли доноров водородных связей, однако их избыток может быть не оптимальным. Наилучшие показатели наблюдаются для соединений с цис-C=C-мостиком (GlideScore ≈ – 8,5 ккал/моль), что подчеркивает важность сохранения планарности и π-сопряжения. Замещение C=C на циклопропановый фрагмент является перспективной стратегией стабилизации цис-конформации и приводит к получению соединений с сопоставимой аффинностью (GlideScore ≈ – 8,2 ккал/моль). Оксадиазольные аналоги в среднем демонстрируют худшие показатели связывания (GlideScore ≈ – 8,0 ккал/моль) и требуют направленной оптимизации геометрии и заместителей. Таким образом, стратегия изостерной модификации молекулярного каркаса за счет введения циклопропанового фрагмента является обоснованной и позволяет получить перспективные соединения, не уступающие эталону по энергии связывания. Несколько соединений из исследованной серии, в частности сохраняющие цис-алкеновый мостик и содержащие комбинацию гидрокси- и метокси-заместителей, продемонстрировали значения GlideScore, превосходящие комбретастатин A-4. Основным ограничением настоящего исследования является узкий выбор предлагаемых модификаций двойной связи, а также предсказательный характер результатов, требующий экспериментальной проверки in vitro. В дальнейшем планируется расширить ряд исследуемых соединений за счет включения других гетероциклических и карбоциклических изостеров, а также провести молекулярно-динамическое моделирование для оценки стабильности комплексов лиганд-белок. Кроме того, перспективным направлением представляется изучение влияния полярности и стерических факторов на проницаемость клеточных мембран и метаболическую стабильность предложенных кандидатов. Полученные результаты имеют практическую ценность для исследователей, занимающихся разработкой противоопухолевых препаратов таргетного действия. Выявленные закономерности «структура–аффинность» и отобранные соединения-кандидаты могут быть использованы для планирования синтеза новых перспективных ингибиторов тубулина и последующего изучения их биологической активности in vitro и in vivo.