Domain Analysis and Visualization of NLRP10

Author: Sim-Hui Tee
Journal: International Journal of Information Technology and Computer Science(IJITCS) @ijitcs
Article in issue: 9 Vol. 5, 2013.
Free access
NLRP10 is one of the members of NOD-like receptors (NLRs) family that is least characterized. It is a protein that takes part in pathogen sensing and responsible for the subsequent signaling propagation leading to immunologic response. In this study, computational tools such as algorithm, web server and database were used to investigate the domain of NLRP10 protein. The findings of this research may provide computational insights into the structure and functions of NLRP10, which in turn may foster better understanding of the role of NLRP10 in the immunologic defense.
Scientific Computing, Bioinformatics, Database, Algorithm, Visualization, Protein, Server
Short address: https://sciup.org/15011963
IDR: 15011963
Text of the scientific article Domain Analysis and Visualization of NLRP10
Published Online August 2013 in MECS
-
I. Introduction
Computer tools are proved indispensable and useful in the research of many scientific fields, especially in advancing human knowledge in the structures and functions of biological processes and entities. In the past decades, various algorithms [1-7], computational models [8-12], web servers [13-18], simulations [1923], databases [24-28], computational intelligence approaches [29-33], and imaging techniques [34-38] have been developed to aid the analysis and process of complex biological data. The use of computational tools has become prevalent in many biomedical niches such as data mining [39-40], sequence analysis [41-43], computational biology [44-49], structural
bioinformatics [50-52], molecular designs [53-55], systems biology [56-58], protein science [59-61], drug discovery [62-63], and even biophysics [64-66]. With these computational tools, biological data can be categorized and analyzed according to the scientific needs. Besides, greater accuracy and structural insights of the molecular data are made possible with computational approaches. Computational solutions to the gigantic volume of biological data appear to be a promising approach to aid the advancement of biological sciences [67].
In this study, we employed computer modelling and fold prediction approach in the study of the domain of NLRP10, which is one of the members of NOD-like receptors (NLRs) family [68]. NLR family members are the main constituents of the inflammasome, which is a molecular assembly that is activated upon infection or cellular stress [68]. Inflammasomes are important in regulating the innate immune defenses by inducing proinflammatory cytokines interleukin-1β and interleukin-18 [68-69]. NLRs have also been recognized to complement the immunological functions of Toll-like receptors (TLRs), a group of pathogen sensors which are membrane-bound and triggering the transcription factor NF-ĸB, mitogen activated protein kinases and Jun amino-terminal kinase [70-72]. Most of the NLRs and TLRs induce the inflammatory response by recruiting adaptor proteins [71-76]. Such recruitment requires the identical or similar structural domain of the interfacing proteins. Hence, the understanding of the protein domain is important for a better insight in the signaling pathways of these pathogen sensing mechanisms. Despite some of the NLRs, such as NLRP3, have been widely studied, the molecular details of NLRP10 remain poorly understood. The understanding of NLRP10, especially its domain, would greatly elucidate the downstream signaling pathways and the immunologic mechanism of cytokine induction.
Domain analysis is an approach which has been widely adopted by bioinformaticians and computer scientists in the analysis of protein structure. The common computational approach in domain analysis lies in the identification of motif, such as those applied in SLiM-mediated protein interactions [77], homology study [78,83], localization of structural motifs [79], and the binding site identification [80-81]. Besides, theoretical computer science, such as the concept of graph theory, is also frequently applied in the analysis of protein domain [82]. Because the structural insights of proteins are closely associated to protein function, domain analysis is vital and versatile in revealing the cellular processes. To date, the known domains of NLRP10 are Nucleotide-binding and Oligomerization (NACHT) and pyrin (PYD). This research undertakes to analyze the domains of NLRP10 using computational tools. The findings of this research may provide computational insights into the structure and functions of NLRP10, which is crucial for further investigation in the fields such as structural bioinformatics and immunopathology.
In this paper, the procedures and methods are described in detail in Section II. The obtained results were presented in Section III with elaborated discussion. A conclusion of the findings is given in Section IV.
-
II. Methods
The nucleotide and amino acid sequence of NLRP10 were retrieved from the National Center for Biotechnology Information (NCBI). We used neural network based Pcons [85] to find the structural templates for NLRP10. Upon the identification of the modelling template for NLRP10, NMR Restraints Grid [84] was used to identify the NMR data. NRG-CING database [86] was used to model and validate the structure of the template for NLRP10. The algorithm used in the structural modelling is Saltbridge [88]. In addition, we used Ramachandran plot [89] to visualize backbone dihedral angles Ψ against φ of amino acid residues in the protein. The backbone of a protein was considered as a discrete curve, which permits the Frenet frames to be calculated based on space curves [87]. Let
We define a unit tangent vector at point P j , where j =0,…, n -1, as such:
+ + 1 - P j
tj
S j
The points of the curve were computed from the translation of the sequences {tj} and {sj} by k-1
k - P0 =S Sjt, j=0
-
III. Results and Discussion
NLRP10 is a protein constituted by 655 amino acids. It has a Pyrin domain at its N-terminus and a central NACHT domain, as depicted in Fig. 1.
S j = p + 1 - pj l
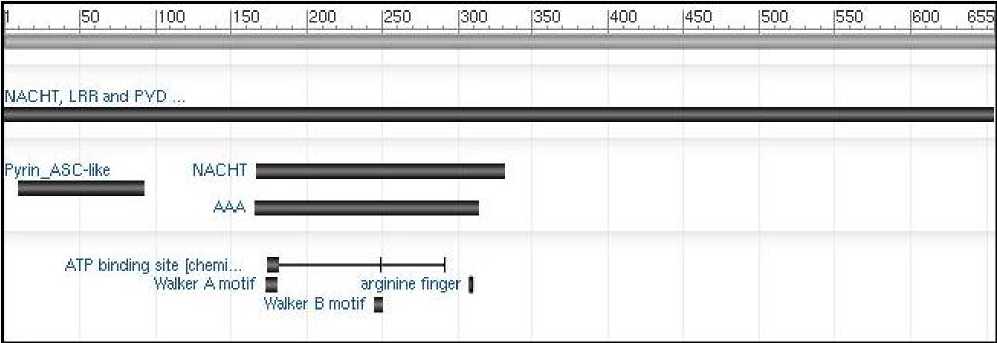
Fig. 1: The schematic view of NLRP10 domains
Using Pcons server [85], we obtained 5 protein templates which serve as structural models for NLRP10. These templates are summarized in Table 1.
Table 1: Protein templates for NLRP10
|
Rank |
Pcons score |
ProQ score |
Template |
|
1 |
0.050 |
117.57 |
2KN6 |
|
2 |
0.047 |
58.01 |
2HM2 |
|
3 |
0.047 |
60.36 |
1UCP |
|
4 |
0.046 |
68.40 |
2DO9 |
|
5 |
0.044 |
56.24 |
1PN5 |
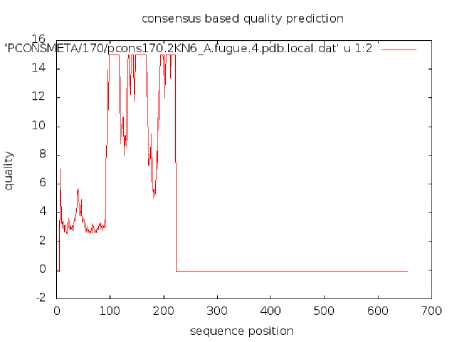
Fig. 2: Consensus based quality prediction for 2KN6
Among 5 candidate proteins, 2KN6 is the best matched protein template with NLRP10, based on the amino acid sequence alignment. The consensus based quality prediction for 2KN6 was depicted in Fig. 2.
It is clear that the predicted quality index is highest between amino acid positions of 100-200 range, though there is a flux in quality index in this range. The predicted quality index is dropped after position 200, drastically, to zero and below. Notably, the second half of the protein exhibits negative values in the consensus based quality prediction. Since sequence consensus does not totally reflect the structure, we performed structural based quality prediction for 2KN6, as illustrated in Fig. 3.
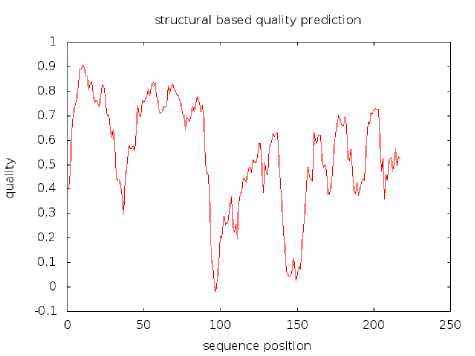
Fig. 3: Structural based quality prediction for 2KN6
Fig 3 demonstrates that the quality prediction based on the structure of 2KN6 has positive quality index (except at position 97, tyrosine), with most parts of the protein acquiring quality index higher than zero. The lowest value is -0.02216 at position 97, following with 0.01278 at position 96 (valine). The highest quality index is 0.90657 at position 10 (tryptophan).
NMR Restraints Grid [84] was used to identify the NMR data for 2KN6. The completeness statistics are summarized in Table 2.
Table 2: The Completeness statistics for 2KN6
|
Parameters |
Values |
|
Model count |
20 |
|
Residue count |
215 |
|
Total atom count |
3060 |
|
Redundancy threshold % |
5.0 |
|
Completeness cutoff |
4.0 |
|
Completeness cumulative % |
36.3 |
|
Constraint unexpanded count |
2495 |
|
Constraint intra-residue count |
793 |
|
Constraint observed count |
1544 |
|
Constraint expected count |
2546 |
|
Constraint matched count |
924 |
|
Constraint unmatched count |
620 |
We queried NRG-CING database [86] to model and validate the NMR data of 2KN6, which is a model template for NLRP10. The side chain and backbone validation are depicted in Fig 4.
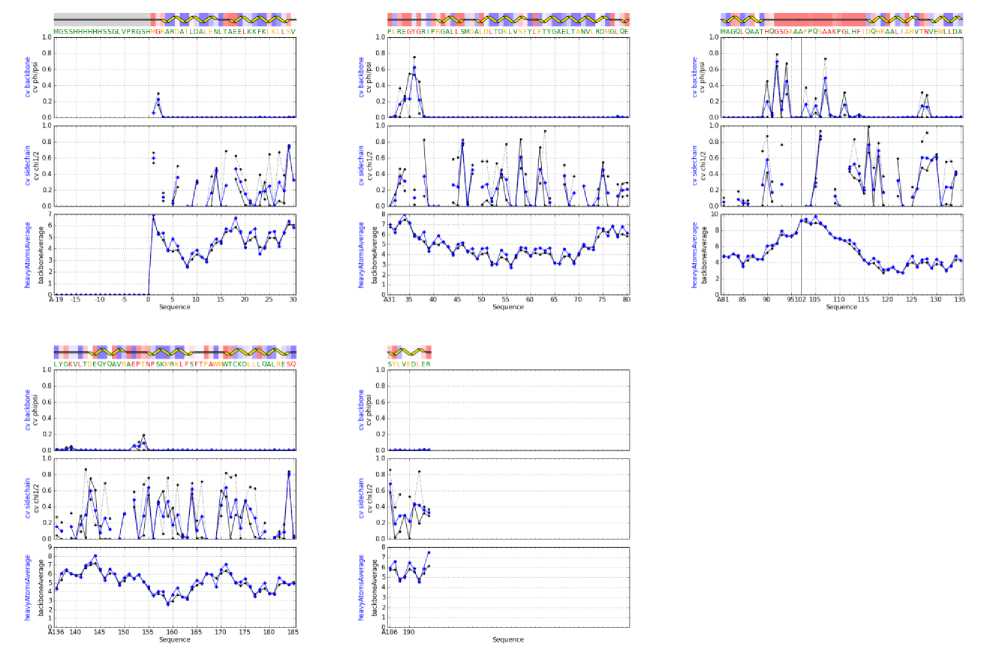
Fig. 4: Side chain and backbone validation
The side chain and backbone validations of 2KN6 NMR data (as shown in Fig 4) demonstrate varieties of structural angles (PHI, PSI, CHI1/2). The angle at certain amino acid position at the side chain is drastically large, as shown at position 28-29, 45-47, 57-59, 115-117, 117-119, and 183-185. The residue properties of 2KN6 were probed. Fig. 5 illustrates one segment of the sequences.
c. RMS devs from mean coords: main-chain (black) and sidechain (grey) 150
oo ll ltllaeHaatlLlllIfctlLillLllllll.il ll I1MII UllWellaal
5 10 15 20 25 30 35 40 45 50

Helix [ - Beta strand wi Random coil АссеамЫйу Moding: ■ Buncd
-
e. Sequence & average estimated accessibilities • AccewNe • Buried lileeeeeeeeeeeeeieееееееееееееееиее•♦•<•••••e<••♦•••<
MGRARDAI LDAL EN I. TAE E L К К F К I. К I I. S V P LREGYGR I PRO A 1. L SMDALDLTI
Fig. 5: The residue properties of 2KN6 (amino acid 0-55)
The top panel in Fig. 5 shows the RMS deviation of the model from the template. To have an accurate model, it is desirable to have a small RMS deviation. Our obtained results show that the deviation is small enough, reflecting an accurate match between model and template. The middle panel in Fig. 5 demonstrates the accessibility of the secondary structure. In overall, it was noticed that a large portion of the sequence demonstrates a pattern of random coil, implying that the protein backbone will sample all possible structures in the absence of stabilized interactions. As shown in Fig. 5, statistical random coil was found in the vicinity of amino acid position 1-3, 15-17, 30-40, and 46-48. Besides, random coils were also identified in the vicinity of the position 60-63, 77-79, 90-115, 126-128, 135-143,150-155, and 167-170 (data not shown). These suggest that non-local amino acid interactions are absent in these random coil regions. The bottom panel of Fig 5 depicts the accessibility of the sequence to solvent and other binding proteins.
To understand the allowable regions of the residue, we have used Ramachandran plot for this purpose. The plot represents each amino acid residue as a dot in a graph of φ against backbone dihedral angles Ψ. The residues in favored region and generously allowed region are shown in red dot and yellow dot, respectively.
From Fig 6, we notice that the residues in favored region are clustered largely negative for φ whereas positive for Ψ. The triangles in Fig 6 represent glycine residues, which provide flexibility for enzyme active sites [90]. We summarized the plot statistics in Table 3.
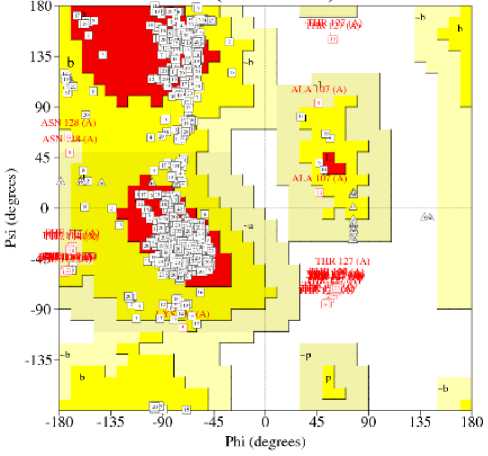
Fig. 6: Ramachandran plot
Table 3: Statistics of Ramachandran plot
|
Residues in most favored regions |
1470 |
86.5% |
|
Residues in additional allowed regions |
198 |
11.6% |
|
Residues in generously allowed regions |
14 |
0.8% |
|
Residues in disallowed regions |
18 |
1.1% |
|
Number of non-glycine and non-proline residues |
1700 |
100% |
|
Number of glycine residues |
40 |
|
|
Number of proline residues |
120 |
|
|
Total number of residues |
1860 |
From Table 3, it is evident that most of the residues are falling within the most favored regions (86.5%). The number of glycine and proline residues is low, with a total percentage of 8.6% of the total number of residues. We obtained the distant restraints based on short-, medium-, and long-ranged sequence separation.
Restraints within the same residues are short-range; sequence separation within four residues is mediumrange; and sequence separation greater than four residues is categorized as long-range restraint. The distant restraint is plotted in the chart as shown in Fig 7.
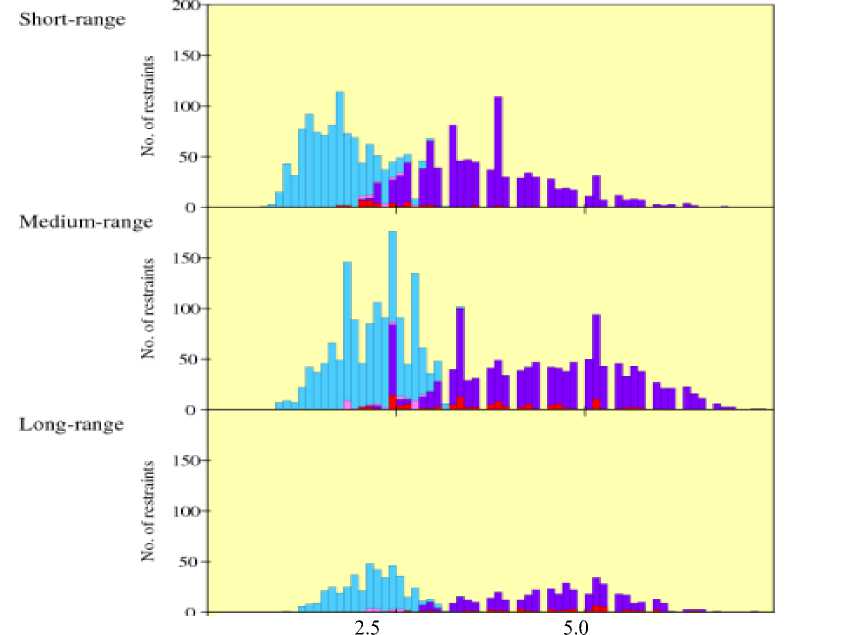
Distance (A)
-
■ Number of upper bound distance restraint» Data subdivided according to:-
- I Number of lower-bound distance restraints Short-range (restraints within same residue)
-
■ Number of upper-hound violation* Medium-range (sequence separation <= 4 residues)
-
■ Number of lower-bound violations Long-range (sequence separation > 4 residues)
Fig. 7: Distant restraints (short-, medium-, and long-range)
As shown in Fig 7, the number of long-range distant restraints reduced drastically (<50) across the residue distance. Regardless of the residue range categories, the number of restraint at smaller distance ( ≤ 2.5 A) is greater than that of greater distance ( ≥ 5.0 A). The data collected on distant restraint is reliable as the number of upper- and lower-bound violations is very low.
position 97, tyrosine), with most parts of the protein acquiring quality index higher than zero. The side chain and backbone of 2KN6 were validated, and the residue properties of 2KN6 were analyzed. Our analysis of the structural properties of 2KN6 casts light on the domain features of the NLRP10 protein, which is critical for the understanding of NLRP10-implicated immunologic diseases.
-
IV. Conclusion
A computational approach combining computer modelling and fold prediction has been used in this research to analyze the domain of NLRP10. 2KN6 serves as a structural template because it is the best matched protein template for NLRP10. In general, the structure of 2KN6 has positive quality index (except at
References Domain Analysis and Visualization of NLRP10
- T. Horváth, J. Ramon, and S. Wrobel, “Frequent subgraph mining in outerplanar graphs,” Data Mining and Knowledge Discovery, Vol. 21, pp. 472-508, 2010.
- K.S. Kim, J.H. Seo, S.H. Ryu, M.H. Kim, and C.G. Song, “Estimation algorithm of the bowel motility based on regression analysis of the jitter and shimmer of bowel,” Computer Methods and Programs in Biomedicine, Vol. 104, pp. 426-434, 2011.
- Y. Teng and T. Zhang, “Three penalized EM-type algorithms for PET image reconstruction,” Computers in Biology and Medicine, Vol. 42, pp. 714-723, 2012.
- R-P. Liang, S-Y. Huang, S-P. Shi, X-Y. Sun, S-B. Suo, and J-D. Qiu, “A novel algorithm combining support vector machine with the discrete wavelet transform for the prediction of protein subcellular localization,” Computers in Biology and Medicine, Vol. 42, pp. 180-187, 2012.
- T. Liu, W. Zhao, L. Tian, R. Wu, “An algorithm for molecular dissection of tumor,” Journal of Mathematical Biology, Vol. 50, pp. 336-354, 2005.
- C.E. Hann, P. Docherty, J.G. Chase, and G.M. Shaw, “A fast generalizable solution method for glucose control algorithms,” Mathematical Biosciences, Vol. 227, pp. 44-55, 2010.
- T. Rausch, S. Koren, G. Denisov, D. Weese, A-K. Emde, A. Döring, and K. Reinert, “A consistency-based consensus algorithm for de novo and reference-guided sequence assembly of short reads,” Bioinformatics, Vol. 25, No. 9, pp. 1118-1124, 2009.
- S. Gunewardena and Z. Zhang, “A hybrid model for robust detection of transcription factor binding sites,” Bioinformatics, Vol. 24, No. 4, pp. 484-491, 2008.
- M.A Jonikas, R.J. Radmer, and R.B. Altman, “Knowledge-based instantiation of full atomic detail into coarse-grain RNA 3D structural models,” Bioinformatics, Vol. 25, No. 24, pp. 3259-3266, 2009.
- S. Loriot and F. Cazals, “Modeling macro-molecular interfaces with Intervor,” Bioinformatics, Vol. 26, No. 7, pp. 964-965, 2010.
- C-F. Huang, J. Kaur, A. Maguitman, and L.M. Rocha, “Agent-based model of genotype editing,” Evolutionary Computation, Vol. 15, pp. 253-289, 2007.
- T. Müller, R. Spang, and M. Vingron, “Estimating amino acid substitution models: a comparison of Dayhoff’s estimator, the resolvent approach and a maximum likelihood method,” Molecular Biology and Evolution, Vol. 19, pp. 8-13, 2002.
- D. Talavera, R.A. Laskowski, and J.M. Thornton, “WSsas: a web service for the annotation of functional residues through structural homologues,” Bioinformatics, Vol. 25, No. 9, pp. 1192-1194, 2009.
- C. Yeats, J. Lees, P. Carter, I. Sillitoe, and C. Orengo, “The Gene3D web services: a platform for identifying, annotating and comparing structural domains in protein sequences,” Nucleic Acids Research, Vol. 39, pp. W546-W550, 2011.
- F. Poitevin, H. Orland, S. Doniach, P. Koehl, and M. Delarue, “AquaSAXS: a web server for computation and fitting of SAXS profiles with non-uniformally hydrated atomic models,” Nucleic Acids Research, Vol. 39, pp. W184-W189, 2011.
- E. Tjioe, K. Lasker, B. Webb, H.J. Wolfson, and A. Sali, “MultiFit: a web server for fitting multiple protein structures into their electron microscopy density map,” Nucleic Acids Research, Vol. 39, pp. W167-W170, 2011.
- J-D. Qiu, S-B. Suo, X-Y. Sun, S-P. Shi, and R-P. Liang, “OligoPred: a web-server for predicting homo-oligomeric proteins by incorporating discrete wavelet transform into Chou’s pseudo amino acid composition,” Journal of Molecular Graphics and Modelling, Vol. 30, pp. 129-134, 2011.
- M. DiBernardo, R. Pottinger, and M. Wilkinson, “Semi-automatic web service composition for the life sciences using the BioMoby semantic web framework,” Journal of Biomedical Informatics, Vol. 41, pp. 837-847, 2008.
- J.H. Gennari, M.L. Neal, M. Galdzicki, and D.L. Cook, “Multiple ontologies in action: composite annotations for biosimulation models,” Journal of Biomedical Informatics, Vol. 44, pp. 146-154, 2011.
- G.D. O’Clock, Y.W. Lee, J. Lee, and W.J. Warwick, “A simulation tool to study high-frequency chest compression energy transfer mechanisms and waveforms for pulmonary disease applications,” IEEE Transactions on Biomedical Engineering, Vol. 57, No. 7, pp. 1539-1546, 2010.
- M-D. Tsai and M-S. Hsieh, “Volume manipulations for simulating bone and joint surgery,” IEEE Transactions on Information Technology in Biomedicine, Vol. 9, No. 1, pp. 139-149,2005.
- S. Costantini, G. Colonna, A.M. Facchiano, “Simulation of conformational changes occurring when a protein interacts with its receptor,” Computational Biology and Chemistry, Vol. 31, pp. 196-206, 2007.
- H. Ogawa, M. Nakano, H. Watanabe, E. Starikov, S.M. Rothstein, and S. Tanaka, “Molecular dynamics simulation study on the structural stabilities of polyglutamine peptides,” Computational Biology and Chemistry, Vol. 32, pp. 102-110, 2008.
- S. Moon, Y. Byun, and K. Han, “FSDB: a frameshift signal database,” Computational Biology and Chemistry, Vol. 31, pp. 298-302, 2007.
- J-T. Horng, F.M. Lin, J.H. Lin, H.D. Huang, and B.J. Liu, “Database of repetitive elements in complete genomes and data mining using transcription factor binding sites,” IEEE Transactions on Information Technology in Biomedicine,” Vol. 7, No. 2, pp. 93-100, 2003.
- D. Taylor, R.N.G. Naguib, and S. Boulton, “A dynamic clinical dental relational database,” IEEE Transactions on Information Technology in Biomedicine, Vol. 8, No. 3, pp. 298-305, 2004.
- V.A. Gennarino, M. Sardiello, M. Mutarelli, G. Dharmalingam, V. Maselli, G. Lago, and S. Banfi, “HOCTAR database: a unique resource for microRNA target prediction,” Gene, Vol. 480, pp. 51-58, 2011.
- F. Piva, M. Giulietti, L. Nocchi, and G. Principato, “SpliceAid: a database of experimental RNA target motifs bound by splicing proteins in humans,” Bioinformatics, Vol. 25, No. 9, pp. 1211-1213, 2009.
- M. Pellegrini, M.E. Renda, and A. Vecchio, “TRStalker: an efficient heuristic for finding fuzzy tandem,” Bioinformatics, Vol. 26, pp. i358-i366, 2010.
- S-Y. Ho, C-H. Hsieh, H-M. Chen, and H-L. Huang, “Interpretable gene expression classifier with an accurate and compact fuzzy rule base for microarray data analysis,” BioSystems, Vol. 85, pp. 165-176, 2006.
- G. Dudek, Z.J. Grzywna, and M.L. Willcox, “Classification of antituberculosis herbs for remedial purposes by using fuzzy sets,” BioSystems, Vol. 94, pp. 285-289, 2008.
- R. Brette, “Generation of correlated spike trains,” Neural Computation, Vol. 21, pp. 188-215, 2009.
- H. Chen, S. Saïghi, L. Buhry, and S. Renaud, “Real-time simulation of biologically realistic stochastic neurons in VLSI,” IEEE Transactions on Neural Networks, Vol. 21, No. 9, pp. 1511-1517, 2010.
- R.I. Ionasec, I. Voigt, B. Georgescu, Y. Wang, H. Houle, F. Vega-Higuera, N. Navab, and D. Comaniciu, “Patient-specific modeling and quantification of the aortic and mitral valves from 4-D cardiac CT and TEE,” IEEE Transactions on Medical Imaging, Vol. 29, No. 9, pp. 1636-1651, 2010.
- J. Duan, S.L. Dixon, J.F. Lowrie, and W. Sherman, “Analysis and comparison of 2D fingerprints: insights into database screening performance using eight fingerprint methods,” Journal of Molecular Graphics and Modelling, Vol. 29, pp. 157-170, 2010.
- E. Yang, P. Liu, D.N. Rassokhin, and D.K. Agrafiotis, “Stochastic proximity embedding on graphics processing units: taking multidimensional scaling to a new scale,” Journal of Chemical Information and Modeling, Vol. 51, pp. 2852-2859, 2011.
- R. Eslami and M. Jacob, “Robust reconstruction of MRSI data using a sparse spectral model and high resolution MRI priors,” IEEE Transactions on Medical Imaging, Vol. 29, No. 6, pp. 1297-1309, 2010.
- F. Yang and T. Jiang, “Cell image segmentation with kernel-based dynamic clustering and an ellipsoidal cell shape model,” Journal of Biomedical Informatics, Vol. 34, pp. 67-73, 2001.
- H.P. Benton, E.J. Want, and T. Ebbels, “Correction of mass calibration gaps in liquid chromatography-mass spectrometry metabolomics data,” Bioinformatics, Vol. 26, No. 19, pp. 2488-2489, 2010.
- Y. Kano, P. Dobson, M. Nakanishi, J. Tsujii, and S. Ananiadou, “Text mining meets workflow: linking U-Compare with Taverna,” Bioinformatics, Vol. 26, No. 19, pp. 2486-2487, 2010.
- T.A. Peterson, A. Adadey, I. Santana-Cruz, Y. Sun, A. Winder, and M.G. Kann, “DMDM: domain mapping of disease mutations,” Bioinformatics, Vol. 26, No. 19, pp. 2458-2459, 2010.
- A. Taneda, “Multi-objective pairwise RNA sequence alignment,” Bioinformatics, Vol. 26, No. 19, pp. 2383-2390, 2010.
- H. Abel, E. Duncavage, N. Becker, J.R. Armstrong, V.J. Magrini, and J.D. Pfeifer, “SLOPE: a quick and accurate method for locating non-SNP structural variation from targeted next-generation sequence data,” Bioinformatics, Vol. 26, No. 21, pp. 2684-2688, 2010.
- X. Meng, Q. Lu, and J. Rinzel, “Control of firing patterns by two transient potassium currents: leading spike, latency, bistability,” Journal of Computational Neuroscience, Vol. 31, No. 1, pp. 117-136, 2011.
- J.D. Drover, N.D. Schiff, and J.D. Victor, “Dynamics of coupled thalamocortical modules,” Journal of Computational Neuroscience, Vol. 28, pp. 605-616, 2010.
- Y-K. Yu and S.F. Altschul, “The complexity of the Dirichlet model for multiple alignment data,” Journal of Computational Biology, Vol. 18, No. 8, pp. 925-939, 2011.
- L-C. Wu, J-T. Horng, and Y-A. Chen, “A computation to integrate the analysis of genetic variations occurring within regulatory elements and their possible effects,” Journal of Computational Biology, Vol. 16, No. 12, pp. 1731-1747, 2009.
- K. Krepkin and J. Costa, “Defining the role of cooperation in early tumor progression,” Journal of Theoretical Biology, Vol. 285, pp. 36-45, 2011.
- J.K. Møller, K.R. Bergmann, L.E. Christiansen, H. Madsen, “Development of a restricted state space stochastic differential equation model for bacterial growth in rich media,” Journal of Theoretical Biology, Vol. 305, pp. 78-87, 2012.
- M. Rueda, V. Katritch, E. Raush, and R. Abagyan, “SimiCon: a web tool for protein-ligand model comparison through calculation of equivalent atomic contacts,” Bioinformatics, Vol. 26, No. 21, pp. 2784-2785, 2010.
- T. Nugent and D.T. Jones, “Membrane protein structural bioinformatics,” Journal of Structural Biology, Vol. 179, pp. 327-337, 2012.
- M. Andronescu, A. Condon, H.H. Hoos, D.H. Mathews, and K. Murphy, “Computational approaches for RNA energy parameter estimation,” RNA, Vol. 16, pp. 2304-2318, 2010.
- B.L. Howard, P.E. Thompson, and D.T. Manallack, “Active site similarity between human and Plasmodium falciparum phosphodiesterases: considerations for antimalarial drug design,” Journal of Computer-aided Molecular Design, Vol. 25, pp. 753-762, 2011.
- M.A. Alam and P.K. Naik, “Molecular modelling evaluation of the cytotoxic activity of podophyllotoxin analogues,” Journal of Computer-aided Molecular Design, Vol. 23, pp. 209-225, 2009.
- K-C. Shih, C-Y. Lin, H-C. Chi, C-S. Hwang, T-S. Chen, C-Y. Tang, and N-W. Hsiao, “Design of novel FLT-3 inhibitors based on dual-layer 3D-QSAR model and fragment-based compounds in silico,” Journal of Chemical Information and Modeling, Vol. 52, pp. 146-155, 2012.
- R.O. Lindén, V-P. Eronen, and T. Aittokallio, “Quantitative maps of genetic interactions in yeast—comparative evaluation and integrative analysis,” BMC Systems Biology, Vol. 5: 45, 2011.
- P. Wei and W. Pan, “Incorporating gene networks into statistical tests for genomic data via a spatially correlated mixture model,” Bioinformatics, Vol. 24, No. 3, pp. 404-411, 2008.
- K.Y. Yip, P. Patel, P.M. Kim, D.M. Engelman, D. McDermott, and M. Gerstein, “An integrated system for studying residue coevolution in proteins,” Bioinformatics, Vol. 24, No. 2, pp. 290-292, 2008.
- C. Huang, R. Zhang, Z. Chen, Y. Jiang, Z. Shang, P. Sun, X. Zhang, and X. Li, “Predict potential drug targets from the ion channel proteins based on SVM,” Journal of Theoretical Biology, Vol. 262, pp. 750-756, 2010.
- R. Wallace, “Protein folding disorders: toward a basic biological paradigm,” Journal of Theoretical Biology, Vol. 267, pp. 582-594, 2010.
- R. Xu and Y. Xiao, “A common sequence-associated physicochemical feature for proteins of beta-trefoil family,” Computational Biology and Chemistry, Vol. 29, pp. 79-82, 2005.
- L. Yao, Y. Zhang, Y. Li, P. Sanseau, and P. Agarwal, “Electronic health records: implications for drug discovery,” Drug Discovery Today, Vol. 16, pp. 594-599, 2011.
- L. Xie, L. Xie, S.L. Kinnings, and P.E. Bourne, “Novel computational approaches to polypharmacology as a means to define responses to individual drugs,” Annual Review of Pharmacology and Toxicology, Vol. 52, pp. 361-379, 2012.
- B.H. Havsteen, “Dynamic analysis of the atomic vibrations of proteins, as exemplified by the binding of myristic acid to human serum albumin,” European Biophysics Journal, Vol. 38, pp. 1029-1034, 2009.
- B.M. Mills and L.T. Chong, “Molecular simulations of mutually exclusive folding in a two-domain protein switch,” Biophysical Journal, Vol. 100, pp. 756-764, 2011.
- G. Heinzelmann, T. Baştuğ, and S. Kuyucak, “Free energy simulations of ligand binding to the aspartate transporter GltPh,” Biophysical Journal, Vol. 101, pp. 2380-2388, 2011.
- E.E. Schadt, M.D. Linderman, J. Sorenson, L. Lee, and G.P. Nolan, “Computational solutions to large-scale data management and analysis,” Nature Reviews Genetics, Vol. 11, pp. 647-657, 2010.
- K. Schroder and J. Tschopp, “The inflammasomes,” Cell, Vol. 140, pp. 821-832, 2010.
- L.M. Rehaume, T. Jouault, and M. Chamaillard, “Lessons from the inflammasome: a molecular sentry linking Candida and Crohn’s disease,” Trends in Immunology, Vol. 31, No. 5, pp. 171-175, 2010.
- M.H. Shaw, T. Reimer, Y-G. Kim, and G. Nuñez, “NOD-like receptors (NLRs): bona fide intracellular microbial sensors,” Current Opinion in Immunology, Vol. 20, pp. 377-382, 2008.
- L. O’Neill, “How toll-like receptors signal: what we know and what we don’t know,” Current Opinion in Immunology, Vol. 18, pp. 3-9, 2006.
- G.N. Barber, “Innate immune DNA sensing pathways: STING, AIMII and the regulation of interferon production and inflammatory responses,” Current Opinion in Immunology, Vol. 23, pp. 10-20, 2011.
- F. van de Veerdonk, M.G. Netea, C.A. Dinarello, and L. Joosten, “Inflammasome activation and IL-1β and IL-18 processing during infection,” Trends in Immunology, Vol. 32, No. 3, pp. 110-116, 2011.
- I.K. Pang and A. Iwasaki, “Inflammasomes as mediators of immunity against influenza virus,” Trends in Immunology, Vol. 32, No. 1, pp. 34-41, 2011.
- Y. Qiao, P. Wang, J. Qi, L. Zhang, and C. Gao, “TLR-induced NF-ĸB activation regulates NLRP3 expression in murine macrophages,” FEBS Letters, Vol. 586, pp. 1022-1026, 2012.
- S.H. Park, M.S. Kyeong, Y. Hwang, S.Y. Ryu, S-B. Han, and Y. Kim, “Inhibition of LPS binding to MD-2 co-receptor for suppressing TLR4-mediated expression of inflammatory cytokine by 1-dehydro-10-gingerdione from dietary ginger,” Biochemical and Biophysical Research Communications, Vol. 419, pp. 735-740, 2012.
- W. Hugo, F. Song, Z. Aung, S-K. Ng, and W-K. Sung, “SLiM on diet: finding short linear motifs on domain interaction interfaces in protein data bank,” Bioinformatics, Vol. 26, No. 8, pp. 1036-1042, 2010.
- K.O. Kopec, V. Alva, and A.N. Lupas, “Homology of SMP domains to the TULIP superfamily of lipid-binding proteins provides a structural basis for lipid exchange between ER and mitochondria,” Bioinformatics, Vol. 26, No. 16, pp. 1927-1931, 2010.
- M. Sarver, C.L. Zirbel, J. Stombaugh, A. Mokdad, and N.B. Leontis, “FR3D: finding local and composite recurrent structural motifs in RNA 3D structures,” Journal of Mathematical Biology, Vol. 56, pp. 215-252, 2008.
- G. Li, T-M. Chan, K-S. Leung, and K-H. Lee, “A cluster refinement algorithm for motif discovery,” IEEE/ACM Transactions on Computational Biology and Bioinformatics, Vol. 7, No. 4, pp. 654-668, 2010.
- Z. Qin, L. McCue, W. Thompson, L. Mayerhofer, C. Lawrence, and J. Liu, “Identification of co-regulated genes through Bayesian clustering of predicted regulatory binding sites,” Nature Biotechnology, Vol. 21, pp. 435-439, 2003.
- T. Przytycka, G. Davis, N. Song, and D. Durand, “Graph theoretical insights into evolution of multidomain proteins,” Journal of Computational Biology, Vol. 13, No. 2, pp. 351-363, 2006.
- N. Song, R.D. Sedgewick, and D. Durand, “Domain architecture comparison for multidomain homology identification,” Journal of Computational Biology, Vol. 14, No. 4, pp. 496-516, 2007.
- J.F. Doreleijers, A.J. Nederveen, W. Vranken, J. Lin, A.M. Bonvin, R. Kaptein, J.L. Markley, and E.L. Ulrich, “BioMagResBank databases DOCR and FRED with converted and filtered sets of experimental NMR restraints and coordinates from over 500 protein PDB structures,” Journal of Biomolecular NMR, Vol. 32, pp. 1-12, 2005.
- J. Lundstrom, L. Rychlewski, J. Bujnicki, and A. Elofsson, “Pcons: a neural-network-based consensus predictor that improves fold recognition,” Protein Science, Vol. 10, pp. 2354-2362, 2001.
- J.F. Doreleijers, W.F. Vranken, C. Schulte, J.L. Markley, E.L. Ulrich, G. Vriend, and G.W. Vuister, “NRG-CING: integrated validation reports of remediated experimental biomolecular NMR data and coordinates in wwPDB,” Nucleic Acids Research, Vol. 40, pp. D519-D524, 2012.
- J.R. Quine, T.A. Cross, M.S. Chapman, and R. Bertram, “Mathematical aspects of protein structure determination with NMR orientational restraints,” Bulletin of Mathematical Biology, Vol. 66, pp. 1705-1730, 2004.
- S. Kumar and R. Nussinov, “Relationship between ion pair geometries and electrostatic strengths in proteins,” Biophysical Journal, Vol. 83, pp. 1595-1612, 2002.
- G. Ramachandran, C. Ramakrishnan, and V. Sasisekharan, “Stereochemistry of polypeptide chain configurations,” Journal of Molecular Biology, Vol. 7, pp. 95-99, 1963.
- B.X. Yan and Y.Q. Sun, “Glycine residues provide flexibility for enzyme active sites,” Journal of Biological Chemistry, Vol. 272, pp. 3190-3194, 1997.
