Enhanced PROBCONS for multiple sequence alignment in cloud computing

Author: Eman M. Mohamed, Hamdy M. Mousa, Arabi E. keshk
Journal: International Journal of Information Technology and Computer Science @ijitcs
Article in issue: 9 Vol. 11, 2019.
Free access
Multiple protein sequence alignment (MPSA) intend to realize the similarity between multiple protein sequences and increasing accuracy. MPSA turns into a critical bottleneck for large scale protein sequence data sets. It is vital for existing MPSA tools to be kept running in a parallelized design. Joining MPSA tools with cloud computing will improve the speed and accuracy in case of large scale data sets. PROBCONS is probabilistic consistency for progressive MPSA based on hidden Markov models. PROBCONS is an MPSA tool that achieves the maximum expected accuracy, but it has a time-consuming problem. In this paper firstly, the proposed approach is to cluster the large multiple protein sequences into structurally similar protein sequences. This classification is done based on secondary structure, LCS, and amino acids features. Then PROBCONS MPSA tool will be performed in parallel to clusters. The last step is to merge the final PROBCONS of clusters. The proposed algorithm is in the Amazon Elastic Cloud (EC2). The proposed algorithm achieved the highest alignment accuracy. Feature classification understands protein sequence, structure and function, and all these features affect accuracy strongly and reduce the running time of searching to produce the final alignment result.
Bioinformatics, Multiple sequence alignment, Protein features, PROBCONS
Short address: https://sciup.org/15016386
IDR: 15016386 | DOI: 10.5815/ijitcs.2019.09.05
Text of the scientific article Enhanced PROBCONS for multiple sequence alignment in cloud computing
Published Online September 2019 in MECS DOI: 10.5815/ijitcs.2019.09.05
Alignment is the process of putting at least two amino acids in the same columns to achieve the maximum level of similarity, this similarity indicates the relationship between sequences [1].
The alignment algorithms, classified into two categories local and global alignment, global uses the entire sequences expand the quantity of matched residues, for example, a Needleman Wunsch algorithm.
F G K S T K Q T G K G | | | | |
F N A T A K S A G K G
But Local algorithms maximize the alignment of similar subregions, for example, Smith-Waterman algorithm.
_________F G K G _________
| | |
_________F G K T _________
Multiple sequence alignment (MSA) is contained more than pairwise sequences. MSA is aligned simultaneously obtained by inserting gaps (-) into sequences [2, 3]. An example of MPSA is presented in figure 1.
|
SI: |
FGKGKC |
SI’: FG K- |
G К C |
|
52: |
FGKFGK — |
—К S2’- F G К F |
G К -GKC |
|
1 kJ 1 \ 1 kJ I \ |
■ UZ- . 1 kJ l\ 1 |
||
|
S3: |
GKGKC |
~К S3’:- G К - |
|
|
S4: |
KFKC |
S4’> - К F |
- КС |
Fig.1. MSA example, with four protein sequences.
To get the ideal protein MSA, there are many MSA methods. MSA methods are classified into dynamic programming (DP) [4] and heuristic [5] as shown in figure 2. DP gives the optimal MSA. The heuristic techniques divide into progressive [6], iterative [7] and probabilistic [8] technique. Heuristic MSA produces an approximate solution.
DP gives the optimal MSA, but it is more time consuming, so DP used to align a few sequences. The most popular algorithm for DP is called divide and conquer algorithm (DCMSA) [9, 10]. DCMSA algorithm is introduced by Stoye, which protein sequences are divided into two regions, then into four regions and therefore to eight regions and so on until the sequences are shorter to be predetermined or considered small enough. For optimal alignment, the subsequences are then aligned and in the last step, the alignment is assemblies. Therefore, aligning multiple long sequences is divided into several smaller alignment tasks. The main problem in the DCMSA algorithm is how to determine the position for cutting of each sequence.
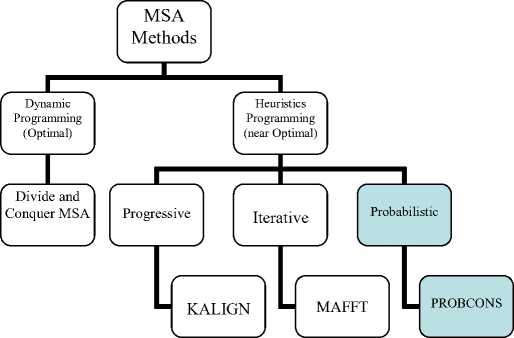
Fig.2. MSA Methods Classification.
The heuristic techniques divide into a progressive, iterative and probabilistic technique. Progressive MSA implemented at three main steps. The first step is calculating the pairwise score and convert them to the distance matrix. The pairwise calculation is done using DP algorithms. The second step is constructing a guide tree from a distance matrix using clustering techniques. Finally, in the last step, align the sequences in their order of the tree. The big advantage of progressive MSA is used to align a large number of biological sequences. However, it produces a near-optimal alignment, which the final alignment depends on the order of aligned pairwise. The most popular programs for progressive MSA is the Clustal family [11, 12] (ClustalX, ClustalW, and Clustal-Omega) and KAlign family (Kalign1, Kalign2, and Kalign-LCS) [13, 14].
Iterative MSA makes an initial alignment of multiple sequences based on some progressive MSA algorithms and then iteratively improve the alignment result to achieve the ideal MSA. For example MAFFT [15] and MUSCLE [16]. MUSCLE tries to make initial MSA as fast as possible and then generate a log-expectation score to perform profile to profile alignment. Unfortunately, previous methods, highly depend on the initial MSA or initial alignment stages.
Finally, for probabilistic procedures, PROBCONS [8] is an MPSA tool that performs progressive consistencybased alignment while representing all imperfect alignment with posterior probability-based scoring. Different highlights of the tool incorporate the utilization of using twofold of affine insertion penalties, guide tree computation through semi probabilistic clustering, iterative refinement, and u nsupervised Expectation-Maximization (EM) preparing of hole parameters. PROBCONS gives a sensational improvement for MPSA accuracy over existing tools. It accomplishes the most astounding scores on the BALIBASE [17] benchmark of any presently realized MPSA tools.
In this paper, clustering the large scale protein sequences based on ten biology protein features. These features classify similar protein sequences to reduce the execution time of PROBCONS tool. Some of the features related to protein secondary structure (PSS) prediction [18, 19]. In order to complete the set of biological features, the classification of an amino acid (AA) is represented [20, 21]. Finally, to achieve accurate alignment, we classify large protein sequences based on the longest common subsequence (LCS) [22]. After that, Apply PROBCONS multiple sequence alignment tools in parallel for each cluster on Amazon EC2 cloud computing platform [23].
The organization of this paper is as follows. Section II reviews the related work of parallelism for large-scale MSA. Section III explains the proposed MPSA. Section IV describes the methods for alignment accuracy measurements and used datasets. The simulation and experimental results are discussed in section V.
-
II. Related work
There are several MSA tools with different attributes, but no single MSA tool can always achieve the highest accuracy with the lowest execution time for all test cases. Parallelization approach is focused to decrease memory and execution time. More different parallelization strategies are implemented to reduce time. Most of the existing MSA parallelization approaches is implemented on multi-core computers [24] or mesh-based multiprocessors [25, 26] or multithreading [27] or MPI (multiprogramming interface) [28] or Hadoop [92] or spark [30] or GPU [31, 32] or clouds [23].
With the rapid growth of biological datasets, MSA techniques must be efficient for large-scale biological data sets. Large-scale MSAs has also the challenge of time and space consuming. Therefore, parallelization is a key approach for decreasing the time execution [33]. There are numerous strategies for alignment with more than two sequences. Some of them minimize time and do not matter by the accuracy of the resulting alignment. Likewise, many strategies maximize accuracy and do not concern with the running time. Decreasing memory and execution time necessities and increasing the MSA accuracy on large-scale datasets are the crucial intention of any technique [33].
In [34] assesses groups with MSA tools of BAliBASE datasets for accuracy, execution time, impacts of sequence length and sequence number. The Results demonstrated that the PROBCONS accuracy is the highest for all the examined MSA tools, yet it was a moderate, slow tool and PROBCONS has no more than 1000 sequences in the alignment.
Classification of protein sequences had an important role in Bioinformatics real-world application. The classification used to understand protein function and protein structure and to know the structure or function of a new protein sequence. In biological research, previous protein classification methods are introduced as different categories [35]. The important methods are feature-based classification and sequence distance based on classification or using HMM [36] or other statistical methods for classification.
Cloud computing is a model that enables flexible computing as a service utility. It provides a scalable infrastructure to compute with storage and other computing issues. There are many cloud providers, which a different service has been offered to users on the internet. Cloud computing has many advantages, which users do not worry about the computing future needs such as maintenance, resources, availability and reliability issues. Cloud users only pay for used resources types and time. As a result, the cloud platform is an important solution for big data analysis, especially in the Bioinformatics research field. Cloud model solves the storage and computational issues for large-scale data analysis [23].
So biology clients don't need to have a high capacity computer for biological data analysis. The cloud provides a high availability data and also provide on-demand powerful computers. The biologists only need internet with high speed to connect with cloud services. For example, Cloud-Coffee [37] is a parallel implementation of T-Coffee but in a different Approach.
PROBCONS (probabilistic-CONSistency-based multiple-alignments-of-amino-acid-sequences) is a tool for creating MPSA dependent on probabilistic consistency. PROBCONS has achieved the most raised accuracy's of all MPSA techniques as of recently. The probabilistic consistency technique has been utilized by PROBCONS for different protein sequences. In any case, PROBCONS cannot be legitimately utilized for multiple template stringing when proteins under thought are distantly-related, which PROBCONS does not utilize much protein structure data in creating a probabilistic MSA; PROBCONS discard gaps-penalty since it is extravagant to appraise the likelihood of a gap. Gappenalty ignoring is good for close protein homologs, but it may affect accuracy when protein sequences are indirectly related. So the fundamental issues in PROBCONS tool is that cannot utilize structure protein data. PROBCONS is not truly fine at distantly-related protein sequence alignment in light of the fact that PROBCONS disregards gap-penalty, so as to accomplish sensible computational productivity [8].
In this paper, PROBCONS is enhanced. It is an MPSA tool that achieves the most expected accuracy, but it has a time-consuming problem. E-PROBCONS is the proposed enhancement of PROBCONS. E-PROBCONS solve the time problem and enlarging the accuracy of the MPSA, which the large multiple protein sequences are clustered into structurally similar protein sequences. Then PROBCONS MPSA tool is performed in parallel on the Amazon Elastic Cloud (EC2).
-
III. Proposed Algorithm
There are various feature protein sequences that may be applied such as Amino-acids classification, average chemical shift [38], k-mer classification [39], and secondary structure prediction. All these features are strongly affected by sequence, accuracy, and similarity.
The fundamental problem with large-scale sequence alignment is time-consuming. Most of current MSA tools are not producing the highest accuracy with less time execution and not suitable for every dataset. MSA with a growing number of sequences (more than 100) is a time consuming and become a big problem to solve. To solve the large-scale problem, the proposed has clustered the protein sequences based on some biological feature. After that, apply PROBCONS MPSA alignment tool in parallel. Finally, merge the alignment results for each cluster as shown in figure 3.
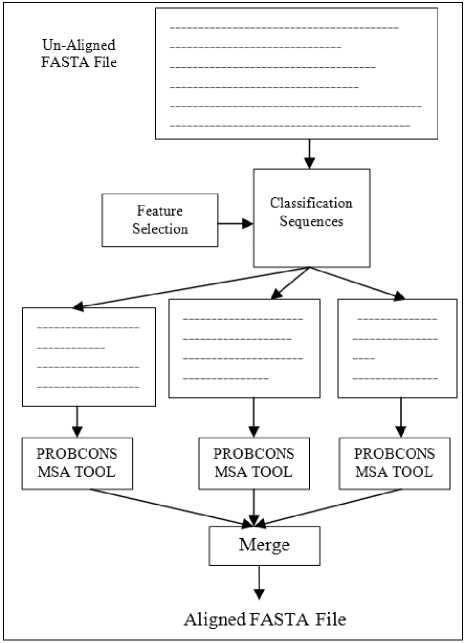
Fig.3. E-PROBCON approach strategy.
Therefore, in first divide protein sequences into groups based on some biological features. So at first let S= S 1 , S 2 , S 3 ,..., S N , which S contains the N Protein sequences.
-
• The sequence S i is placed in the first group. Let M be the group number and Si belongs as a center sequence in M group.
-
• Then, the second sequence S j is compared based on some biological features to S i .
-
• The second sequence would belong to the M group if the result is more than a threshold;
-
• Otherwise, it would form a new group.
-
• For each group, apply a PROBCONS MSA tool.
-
• Merge between groups progressively to retrieve MSA.
-
• Store MSA as FASTA file.
Protein sequences may have similar functions and structures, so in this case, it has high similarity. So in feature selection phase classifying large protein sequences based on the LCS, related to PSS prediction (β- strand, α-helix, and coil structures), and related to amino acid (AA) representation (Aromatic AA, Basic KR AA, Nonpolar AA, Negative Polar Charged AA, Positive Polar Charged AA, and Polar UN Charged AA). The list of features represented in Table 1.
Table 1. Summary of features names that used
|
Feature Name |
|
|
Related to Sequence FLCS |
Number Of Sequences |
|
Average Length |
|
|
Reference Subset |
|
|
Data Type (DNA, Protein) |
|
|
Longest Common Subsequence |
|
|
Related to Secondary Structure FSS |
α-Helix |
|
Β- Sheet |
|
|
Coil |
|
|
Related to Amino Acids FAA |
Polar Uncharged Amino Acids |
|
Nonpolar Aliphatic Amino Acids |
|
|
Basic Positively Charged Amino Acids |
|
|
Aromatic Amino Acids |
|
|
Negatively Charged Amino Acids |
|
|
BasicKR |
|
|
Related to Transmembrane |
Cytoplasmic |
|
Non-Cytoplasmic |
|
|
Transmembrane |
|
|
Related to Average Chemical Shift |
13Cα |
|
13Cβ |
|
|
13C' |
|
|
1Hα |
|
|
1HN |
|
|
15N |
-
• Classification related to the sequence
This feature clustering based on LCS length between two different protein sequences. We define the similarity using LCS for Si and S j as follows:
0 if ( i — j — 0)
LCS ( i , j ) 4
LCS(i -1, j -1) +1 if at — bj max(LCS(i, j -1, LCS(i -1, j)) otherwise
LCS length ( S i , S j ) S — 100
Averagelength ( Si , Sj )
-
• Amino-acids Classification
The amino acid is composed of the 20 amino-acid types. We classify the amino acids as Polar Uncharged Amino Acids ( PUAA ) [G, A, P, V, L, I, M] Percentage, Nonpolar Aliphatic Amino Acids ( NPUAA ) [S, T, C, N, Q] Percentage, Basic Positively Charged Amino Acids ( PCAA ) [K, R, H] Percentage, Aromatic Amino Acids ( AAA ) [F, W, Y] Percentage, Negatively Charged Amino Acids ( NCAA ) [D, E] Percentage and Basic KR residues ( BKR ) [K, R] Percentage as shown in figure 4 and figure 5.
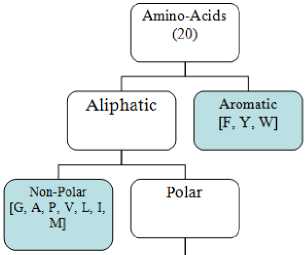
Un-Charged [C, N, Q, S, T]
Charged
Negative Charged FD.E]
Positive Charged Ri, K. RI
Fig.4. Amino-Acids classification.
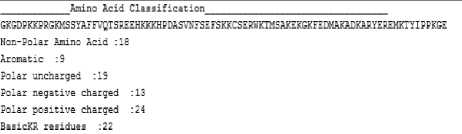
Fig.5. Amino-Acids classification, protein sequence example.
Amino-acids classification is clustered into six categories to reflect the information of sequence order and accuracy based on Amino-acids.
Z n pl = (PUAA) = -=^- (3)
Where ( PUAA ) i is the total percentage of Polar uncharged amino acid type and n i is the counting number of [S, T, C, N, Q] occurring in a protein with sequence length L .
Z ni p 2 = (NPUAA) = ^1— (4)
Where ( NPUAA ) i is a percentage of nonpolar aliphatic amino acid type and n i is the counting number of [G, A, P, V, L, I, M] occurring in a protein with sequence length L .
Z n p 3 = (PCAA ),= (5)
Where ( PCAA )i is the total percentage of positively charged amino acid type and n i is the counting number of [K, R, H] occurring in a protein with sequence length L .
Z ni p 4 = (AAA ),= -=1— (6)
Where (AAA)i is the total percentage of Aromatic amino acid type and ni is the counting of [F, W, Y] occurring in a protein with sequence length L .
Z n p 5 = (NCAA )i = ^=1— (7)
Where ( NCAA ) i is the percentage of negatively charged amino acid type and ni is the number of [D, E] occurring in a protein with sequence length L.
Z n p 6 = (PKR )i = (8)
Where ( BKR ) i is the percentage of basic KR residues amino acid type and n i is the number of [K, R] type occurring in a protein with sequence length L.
Amino-acids features represented in a six-dimensional vector which:
FAA = [p1, p2, p3, p4, p5, p6]
Finally, we used the Euclidian distance between Si, S j to identify the closest-matching based on Amino-acids classification.
EDAA
( pl - p l j )2 + ( p 2 - p 2 j ) 2 +
( p 3 . - p 3 j )2 + ( p 4 . - p 4) + (9)
\ ( p 5 , - p 5 j ) 2 + ( p 6 - p 6 j ) 2
-
• Classification related to the secondary structure:
Protein structure is very important to understand protein function. Protein structure has three main levels of protein structure: primary, secondary, and tertiary as explained in figure 6. The primary structure is the simplest level of protein structure which is the sequence of amino acids.
For example, in figure 7 to GOR IV [40] software result. Figure 8 represents an example of a Secondary structural classification of the protein sequence. For PSS prediction, one of the most widely used tools is the DSSP (Dictionary of Protein Secondary Structure) package [41]. The program gives the predicted secondary structure, h=helix, e=extended or beta-strand, and c=coil; protein structure data can be obtained from protein data bank (PDB).
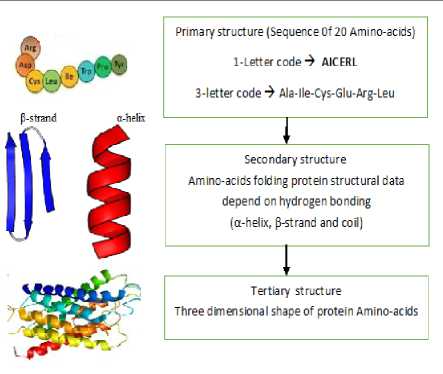
Fig.6. Protein structure levels
PSS has three structural domains α-helix, β-strand, and coil. GOR software is one of the PSS prediction methods. GOR version IV is used to predict protein secondary structure [29].
10 20 30 40 50
I I I I I
GKGDPKKPRGKMSSYAFFVQTSREEHKKKHPDASVNFSEFSKKCSERWKT cccccccccccccceeeeehhhhhhhhhcccccceeeceeccecchhhhh
MSAKE KGKFEDMAKADKARYE REMKTYIPPKGE hchhhhhhhhhhhhhhhhhhhhhhheeecceec
Fig.7. GOR IV Secondary structure prediction result.
Extended strand (Ее) Count in percentage 19.27710843373494 %
Random Coil Count in percentage 34.93975903614458 %
Alfa helix Count in percentage 45.78313253012048 %
Fig.8. Secondary structure classification, protein sequence example.
The knowledge of protein structure will be increasing the accuracy and reduce the time for searching to produce the alignment result and provide the protein function information.
|
n |
||
|
Pai, : |
= £^_ L |
(10) |
|
P p, : |
’-_■ L |
(11) |
|
n |
||
|
pc i |
c L |
(12) |
-
IV. Accuracy Evaluation and Used Datasets
There are many standard techniques for measuring accuracy for the MSA or compare between alignment results. In this paper, SPsore is used for measuring accuracy. SPsore (sum of pairs score) is calculated the sum of the score for each pair in every column of MSA result and compared with the sum of pairs score for MSA reference.
SPscore ( X ,
, X n ) =
£ s ( X , X j )
i . j
E Sr i, j
Where nα is the h counting number, nβ is the number of e and nc total of c numbers in a protein sequence with sequence length L . Secondary structure features represented in a 3-dimensional vector which:
FSS = [pα, pβ, pc]
We use the Euclidean distance for secondary structure between Si , S j to identify the closest-matching based on secondary structure as follows:
EDs-s, = V ( a- a ) + ( P - P j ) + ( C i - C j ) 2 (13)
Which αi is a percentage of α-helix to Si , βi is a percentage of β- Sheet to S i , C i is a percentage of the coil in S i , α j is a percentage of α-helix to S j , β j is a percentage of β- Sheet to S j and C j is a percentage of the coil in S j .
Where S r is the dataset reference score and S(X i , X j ) score between pairwise sequences Xi and X j .
To measure the quality of MSA, there are many protein benchmarks. The used protein data sets in the evaluation are called BAliBASE. In BAliBASE [17], the input sequences and reference alignment available in FASTA format. The comparison is done by computing SPscore between the final alignment of input sequences and reference alignment.
In table 2, six cases are used from BAliBASE v.3. In this paper, to compute SPscore, we used MSA comparator software (MQAT version 2.0.1). Which it allows comparing between alignment reference file and more test alignments (>21MB size). MSA comparator is more efficient than the BAliBASE C program [42].
Table 2. Six cases for evaluation
|
BALIBASE |
Filename |
Seq # |
Average length |
Seq identity |
|
RV11 |
BB11001 |
4 |
86 |
<20% identity |
|
RV12 |
BB12043 |
34 |
318 |
20-40% identity |
|
RV20 |
BB20040 |
87 |
482 |
Up to 3 orphans |
|
RV30 |
BB30003 |
142 |
407 |
<25% residue identity |
|
RV40 |
BB40049 |
62 |
862 |
Up to 400 residues |
|
RV50 |
BB50006 |
60 |
642 |
Up to 100 residues |
-
V. Simulation Results and Discussion
All programs were run under the same environment in the cloud platform. The Amazon EC2 cloud platform is used, all programs run in extra-large 4 CPU – 15 GB – 64 bit and Amazon S3 for storage data with cloud Amazon EC2. For performance evaluation, SPscore accuracy, performance measurement is used in equation 14.
At first, we evaluate for each feature the accuracy performance. The proposed has six features for Aminoacids, namely FPUAA, FNPUAA, FPCAA, FAAA, FNCAA, and FBKR. It has three features for secondary structure, namely Fα, Fβ, and Fc. Secondly, combine the feature by using Euclidian distance formula for Aminoacids features, namely FAA and combine three secondary structure features namely FSS. The proposed has FLCS for the longest common subsequence. Finally, combine the three basic features FLCS, FAA and FSS. After that, apply PROBCONS MSA tool in parallel for each cluster. To return the final alignment, merge the alignment result for all clusters. As explained in figure 9.
The SPscore for measurement accuracy for all different feature sets classifications appears in Table 3. In Table 3, we ranked the set of features based on average SPscore for six test cases. The results presented that Amino-acid classification is affected by accuracy results than without classification. The LCS feature increasing the accuracy with PROBCONS tools for MSA. The results show that the combination of the set of listed feature is affected by the quality of final result alignment.
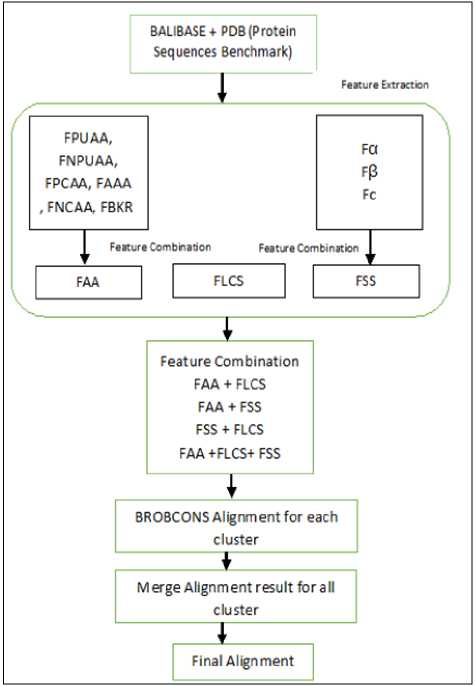
Fig.9. A general framework for E-PROBCONS for large scale dataset
The classification stage partitions the large set file into smaller subgroups to reduce time and we can use PROBCONS tool. PROBCONS has a big problem since the maximum sequence number is limited to 1000 protein sequences. The proposed solved PROBCONS limitation problem. It partitioned the large file into smaller files. The clusters of this file based on some features. As shown in table 3, figure 10 and figure 11, Fα achieves the highest accuracy, followed by the combination of all features. Finally, for execution time evaluation, we compare between PROBCONS and KALIGN without classification, FLCS with PROBCONS, FSS with PROBCONS, FAA with PROBCONS and combination between FAA, FSS and FLCS with PROBCONS as shown in figure 11. The proposed algorithm achieved the highest alignment accuracy. Feature classification understands protein sequence, structure and function, and all these features affect accuracy strongly and reduce the running time of searching to produce the final alignment result.
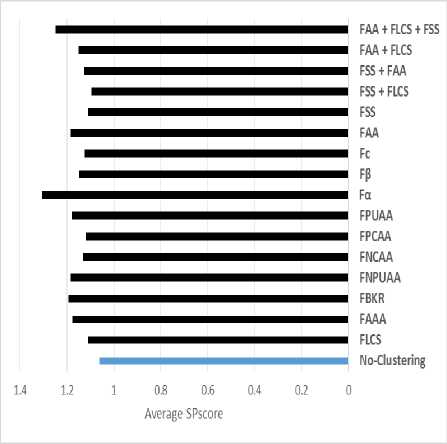
Fig.10. Average SPscore result for PROBCONS with classification or without.
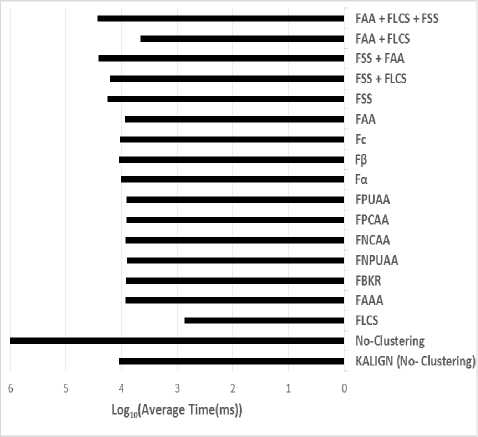
Fig.11. Average execution time for alignment of KALIGN without classification and PROBCONS with classification
Table. 3. The SPscore for accuracy based on different feature classification
|
BAliBASE Benchmark |
||||||||
|
Clustering |
Alignment |
BB11001 |
BB12043 |
BB20040 |
BB30003 |
BB40049 |
BB50006 |
Rank |
|
----- |
KALIGN |
0.482 |
0.965 |
2.226 |
0.658 |
0.478 |
0.747 |
---- |
|
---- |
PROBCONS |
0.542 |
1.063 |
2.362 |
0.848 |
0.656 |
0.903 |
---- |
|
FLCS |
PROBCONS |
0.723 |
1.082 |
2.368 |
0.888 |
0.672 |
0.931 |
11 |
|
FAAA |
PROBCONS |
1.084 |
1.072 |
2.332 |
0.97 |
0.707 |
0.902 |
6 |
|
FBKR |
PROBCONS |
1.084 |
1.138 |
2.373 |
0.942 |
0.735 |
0.892 |
2 |
|
FNPUAA |
PROBCONS |
1.084 |
1.087 |
2.356 |
0.961 |
0.67 |
0.942 |
4 |
|
FNCAA |
PROBCONS |
0.723 |
1.103 |
2.376 |
0.968 |
0.74 |
0.886 |
8 |
|
FPCAA |
PROBCONS |
0.723 |
1.103 |
2.356 |
0.93 |
0.719 |
0.894 |
10 |
|
FPUAA |
PROBCONS |
1.084 |
1.101 |
2.353 |
0.965 |
0.655 |
0.924 |
5 |
|
Fα |
PROBCONS |
0.723 |
1.132 |
2.374 |
1.036 |
1.668 |
0.903 |
1 |
|
Fβ |
PROBCONS |
0.723 |
1.205 |
2.359 |
0.93 |
0.672 |
0.903 |
7 |
|
Fc |
PROBCONS |
0.723 |
1.063 |
2.374 |
0.942 |
0.668 |
0.879 |
9 |
|
FAA |
PROBCONS |
1.084 |
1.142 |
2.366 |
0.955 |
0.665 |
0.89 |
3 |
|
FSS |
PROBCONS |
0.542 |
1.127 |
2.359 |
0.94 |
0.724 |
0.87 |
12 |
|
FSS + FLCS |
PROBCONS |
0.542 |
1.08 |
2.36 |
0.95 |
0.709 |
0.851 |
--- |
|
FSS + FAA |
PROBCONS |
0.723 |
1.176 |
2.359 |
0.848 |
0.722 |
0.933 |
--- |
|
FAA + FLCS |
PROBCONS |
0.542 |
1.141 |
2.371 |
1.036 |
0.898 |
0.918 |
--- |
|
FAA + FLCS + FSS |
PROBCONS |
1.446 |
1.083 |
2.355 |
1.036 |
0.677 |
0.903 |
--- |
-
VI. Conclusion
PROBCONS is a multiple protein sequence alignment (MPSA) tool that achieves the most expected accuracy, but it has a time-consuming problem. To solve this problem and enlarging the accuracy of the MPSA, cluster the large multiple protein sequences into structurally similar protein sequences. Then PROBCONS MPSA tool will be performed in parallel on the Amazon Elastic Cloud (EC2). The Cloud platform is used to reduce the execution time for PROBCONS tool. The maximum accuracy is based on the combination of the protein biological features and classification of the large-scale multiple protein sequences. In this paper, the proposed algorithm using some protein sequence features (LCS, Amino-acids, Secondary structure). The proposed approach is more suitable for large-scale data and shorter sequences. The highest alignment accuracy is achieved and reduce the execution time for producing the alignment result. Comparing with state-of-the-art algorithms (e.g., BROPCONS, and KALIGN), provided more than 50% improvement in terms of average SP score and comparable execution time. The proposed approaches are implemented on the cloud platform in order to improve the scalability with different protein datasets. In future work, we will develop a model using transmembrane classification and average chemical shift factors.
References Enhanced PROBCONS for multiple sequence alignment in cloud computing
- Do CB, Katoh K, " Protein multiple sequence alignment methods" Mol Biol Clifton NJ2008, Vol. 484, pp. 379–413, 2008.
- M. a. Aniba, " Issues in bioinformatics benchmarking: the case study of multiple sequence alignment" Nucleic Acids Res, Vol. 38, pp. 7353–7363, 2010.
- Wallace IM, Blackshields G, Higgins DG., "Multiple sequence alignments" Current Opinion in Structural Biology, Vol. 15, no. 3, pp. 261-266, 2005.
- S. B. Needleman and C. D. Wunsch." A general method Applicable to the Search for Similarities in the Amino Acid Sequence of Two Proteins". Journal of Molecular Biology, Vol. 48(3), pp. 443-453, 1970.
- Peng Zhao, Tao Jiang." A heuristic algorithm for multiple sequence alignment based on blocks". Combinatorial Optimization, Vol. 5(1), pp. 95–115, Mar 2001.
- Feng DF, Doolittle RF., "Progressive sequence alignment as a prerequisite to correct phylogenetic trees", Journal of Molecular Evolution, Vol. 4(25), pp. 351-360, 1987.
- P.Zhao and Tao Jiang J, Hirosawa, M., Totoki, Y., Hoshida, M. and Ishikawa, M., “Comprehensive study on iterative algorithms of multiple sequence alignment”, CABIOS, Vol. 11, pp. 13–18, 1995.
- Chuong B. Do, Mahathi S.P. Mahabhashyam, Michael Brudno, and Serafim Batzoglou, "ProbCons: probabilistic consistency-based multiple sequence alignment", Genome Research, Vol. 2(15), pp. 330-340, 2005.
- Stoye J, "Multiple sequence alignment with the divide-and-conquer method", Gene 211, pp. GC45–GC56, 1998.
- Stoye J, Moulton V, Dress AW, " DCA: an efficient implementation of the divide-and-conquer approach to simultaneous multiple sequence alignment", Comput. Appl. Biosci. Vol.13 (6), pp. 625-626, 1997.
- Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. "Clustal W and Clustal X version 2.0.", Bioinformatics, Vol. 23, pp. 2947-2948, 2007.
- Sievers F, Higgins DG, "Clustal Omega for making accurate alignments of many protein sciences". Protein Sci. , Vol. 27, pp. 135-145, 2018.
- Lassmann T, Sonnhammer EL., "Kalign—an accurate and fast multiple sequence alignment algorithm", BMC Bioinformatics, Vol. 6, pp. 298, 2005.
- Lassmann T, Frings O, Sonnhammer EL." Kalign2: high-performance multiple alignments of protein and nucleotide sequences allowing external features". Nucleic Acids Res, Vol.37, pp. 858–865, 2009.
- Katoh K, Standley DM, "MAFFT multiple sequence alignment software version 7: improvements in performance and usability", Molecular Biology and Evolution, Vol. 4(30), pp. 772–780, 2013
- Edgar RC, "MUSCLE: a multiple sequence alignment methods with reduced time and space complexity", BMC Bioinformatics, Vol. 5, pp. 113-131, 2004.
- Thompson JD, Koehl P, Ripp R, Poch O., "BAliBASE 3.0: latest developments of the multiple sequence alignment benchmark", Proteins, Vol. 1(61), pp. 36-127, 2005.
- Jiang, Q., Jin, X., Lee, S.-J., & Yao, S. "Protein secondary structure prediction: A survey of the state of the art". Journal of Molecular Graphics and Modelling, Vol. 76, pp. 379–402, 2017.
- D.T. Jones, "Protein secondary structure prediction based on position-specific scoring matrices", J. Mol. Biol. Vol. 292, pp. 195–202, 1992.
- Z.-H., Zhou, M., Luo, X., & Li, S. "Highly Efficient Framework for Predicting Interactions between Proteins". IEEE Transactions on Cybernetics, Vol 47(3), pp. 731–743, 2017.
- H. Nakashima, K. Nishikawa, and T. Ooi, “The folding type of a protein are relevant to the amino acid composition,” J. Biochem., Vol. 99(1), pp. 153–162, 1986.
- Bergroth, L., Hakonen, H. and Raita, T. "A Survey of Longest Common Subsequence Algorithms". SPIRE (IEEE Computer Society), pp. 39–48, 2000.
- Daugelaite, J., O’ Driscoll, A., & Sleator, R. D. (2013)." An Overview of Multiple Sequence Alignments and Cloud Computing in Bioinformatics". ISRN Biomathematics, pp. 1–14, 2013.
- Zhu X, Li K, Salah A." A data parallel strategy for aligning multiple biological sequences on multi-core computers". Computers in Biology and Mediciene, Vol. 43(4), pp. 350-361, 2013.
- Charu Sharma and A.K.Vyas "Parallel Approaches in Multiple Sequence Alignments". International Journal of Advanced Research in Computer Science and Software Engineering, Vol. 4(2), 2014.
- Diana H.P.Low, BharadwajVeeravalli, David A.Bader, "On the Design of High-Performance Algorithms for Aligning Multiple Protein Sequences on Mesh-Based Multiprocessor Architectures'' Journal of Parallel and Distributed Computing, no. 67(9), pp. 1007-1017, 2007.
- Chaichoompu K, Kittitornkun S, and Tongsima S. "MT-ClustalW: multithreading multiple sequence alignment"; Proceedings 20th IEEE International Parallel & Distributed Processing Symposium. IEEE Computer Society Press; pp. 280, 2006.
- Kuo-Bin Li. "ClustalW-MPI: ClustalW analysis using distributed and parallel computing". Bioinformatics. ; Vol.19, pp.1585–1586, 2003.
- Quan Zou, Qinghua Hu, Maozu Guo, Guohua Wang. "HAlign: Fast Multiple Similar DNA/RNA Sequence Alignment Based on the Centre Star Strategy". Bioinformatics, Vol. 31(15), pp. 2475-2481, 2015.
- Shixiang Wan, Quan Zou. "HAlign-II: efficient ultra-large multiple sequence alignment and phylogenetic tree reconstruction with distributed and parallel computing". Algorithms for Molecular Biology, pp. 12-25, 2017.
- Blazewicz, J., Frohmberg, W., Kierzynka, M., Wojciechowski, P. "G-MSA - A GPU-based, fast and accurate algorithm for multiple sequence alignment ". Journal of Parallel and Distributed Computing, Vol. 73(1), pp. 32–41, 2013.
- Xi Chen, Chen Wang, Shanjiang Tang, Ce Yu, Quan Zou. "CMSA: A heterogeneous CPU/GPU computing system for multiple similar RNA/DNA sequence alignment". BMC Bioinformatics, Vol. 18, pp. 315, 2017.
- Kleinjung J, Douglas N, Heringa J. "Parallelized multiple alignments". Bioinformatics, Vol.18, pp. 1270–127, 2002.
- Eman M. Mohamed, Hamdy M. Mousa, Arabi E. Keshk, “comparative analysis of multiple sequence alignment tools", MECS, Vol. 10(8), pp. 24-30, 2018.
- Xing, Z., Pei, J., & Keogh, E." A brief survey on sequence classification". ACM SIGKDD Explorations Newsletter, Vol. 12(1), pp. 40, 2010.
- Y. Altun, I. Tsochantaridis, and T. Hofmann. "Hidden Markov support vector machines". ICML '03, the Twentieth International Conference on Machine Learning, pp. 3 -10, 2003.
- Paolo Di Tommaso, Miquel Orobitg, Fernando Guirado, Fernado Cores, Toni Espinosa, Cedric Notredame, " Cloud-Coffee: implementation of a parallel consistency-based multiple alignment algorithm in the T-Coffee package and its benchmarking on the Amazon Elastic-Cloud.," Bioinformatics, Vol. 15(26), pp. 1903-1904, 2010.
- S.P. Mielke, V.V. Krishnan, "Protein structural class identification directly from NMR spectra using averaged chemical shifts", Bioinformatics. Vol. 19, pp. 2054–2064, 2003.
- J. Kähärä and H. Lähdesmäki, “Evaluating a linear k-mer model for protein–DNA interactions using high-throughput SELEX data,” BMC Bioinformat., vol. 14(10), pp. S2, 2013.
- Sen TZ, Jernigan RL, Garnier J, Kloczkowski A. "GOR V server for protein secondary structure prediction". Bioinformatics. Vol. 21(11), pp. 2787–2788, 2005.
- Kabsch W, Sander C. "A dictionary of secondary structure." Biopolymers; Vol. 22, pp. 2577–2637, 1983.
- Pervez MT, Babar ME, Nadeem A, et al. "IVisTMSA: Interactive Visual Tools for Multiple Sequence Alignments". Evol Bioinform Online. Vol. 11, pp.35–42, 2015.
