Фрагментация фосфопептидов методом ECD / ETD в современной тандемной масс-спектрометрии

Автор: Краснов Илья Александрович, Гаврик М.А., Подольская Е.П., Краснов Н.В.
Журнал: Научное приборостроение @nauchnoe-priborostroenie
Рубрика: Масс-спектрометрия
Статья в выпуске: 1 т.23, 2013 года.
Бесплатный доступ
В работе изложены возможности и указаны области применения методов фрагментации пептидов в тандемной масс-спектрометрии, в особенности фрагментации фосфопептидов методами CID, ECD и ETD, а также их комбинациями. Проанализированы механизмы образования фрагментов, методики пробоподготовки и ввода пробы применительно к методу ECD/ETD. Показана возможность использования общих баз данных по фрагментам пептидов для решения задач фосфопротеомики. Сделан вывод об эффективности применения комбинации методов фрагментации CID/ETD для расширения возможностей по восстановления аминокислотных последовательностей фосфопептидов.
Масс-спектрометрия, тандемная масс-спектрометрия, источник ионов, фрагментация, фосфопептиды, протеомика
Короткий адрес: https://sciup.org/14264843
IDR: 14264843 | УДК: 543.51;
Method ECD / ETD fragmentation of the phosphopeptides in modern tandem mass spectrometry
The paper presents features and the application of peptides fragmentation method in tandem mass spectrometry, in particular fragmentation techniques CID, ECD and ETD, and their combinations. Mechanism of fragmentation, sample preparation methods and sample introduction in relation to the method of ECD / ETD are analyzed. The possibility to use common proteomic databases for solving phosphoproteomic tasks is shown. The conclusions about the effectiveness of CID / ETD fragmentation methods combination to expand amino acid sequence restore capacity of the phosphopeptides are made.
Текст обзорной статьи Фрагментация фосфопептидов методом ECD / ETD в современной тандемной масс-спектрометрии
В работе изложены возможности и указаны области применения методов фрагментации пептидов в тандемной масс-спектрометрии, в особенности фрагментации фосфопептидов методами CID, ECD и ETD, а также их комбинациями. Проанализированы механизмы образования фрагментов, методики пробоподготовки и ввода пробы применительно к методу ECD/ETD. Показана возможность использования общих баз данных по фрагментам пептидов для решения задач фосфопротеомики. Сделан вывод об эффективности применения комбинации методов фрагментации CID/ETD для расширения возможностей по восстановления аминокислотных последовательностей фосфопептидов.
Кл. сл. : масс-спектрометрия, тандемная масс-спектрометрия, источник ионов, фрагментация, фосфопептиды, протеомика
ВВЕДЕНИЕ фрагментации. Для фрагментных ионов пептидов принята классификация, предложенная в 1984 г.
Масс-спектрометрия на сегодняшний день является одним из наиболее востребованных инструментальных методов анализа во многих областях науки. Масс-спектрометры (МС) как основной инструмент метода нашли самое широкое применение во многих областях современных наук — химии (биохимия, нефтехимия, аналитическая химия), фармацевтике, медицине, криминалистике, металлургии, защите окружающей среды и многих других. Широкое применение масс-спектрометрия нашла в различных аспектах "науки о жизни" (life science), которая включает в себя технологические аспекты молекулярной биологии и биотехнологии, что хорошо представлено в целом ряде книг и обзоров, например [1, 2].
Отдельно можно выделить тандемные масс-спектрометры (МС-МС), состоящие из двух или более масс-спектрометров, которые широко применяются в области протеомики [3]. В каждый момент времени из всех ионов, образующихся в источнике, для последующей фрагментации отбираются ионы только с одного соединения с заданным отношением массы к заряду. Такая селекция производится при помощи специально разработанных алгоритмов (Data Dependent Acquisition, DDA) на основе обработки данных масс-спектра родительских ионов. Данные одного МС-МС-анализа, как правило, содержат множество фраг-ментных масс-спектров, каждый из которых характеризует фрагменты только одного соединения. Состав фрагментов отражает структуру анализируемого вещества и напрямую зависит от способа в работе [4], в соответствии с которой фрагменты, содержащие N-концевую аминокислоту, в зависимости от разорванной связи обозначают латинскими буквами a, b и c. Подобно этому фрагменты C-конца обозначены x, y и z.
Несмотря на успехи тандемной МС в области протеомики, классические методы фрагментации пептидных ионов зачастую не позволяют получать полную информацию о посттрансляционных модификациях (ПТМ) белков и пептидов. Именно эта информация в настоящее время становится важной для исследований многих биологических процессов. В частности, фосфорилирование белков регулирует межклеточные взаимодействия, пролиферацию, дифференциацию и апоптоз клеток. Последствиями дефектов в процессах фосфорилирования-дефосфорилирования часто становятся различные заболевания, что также повышает важность расширения понимания процессов фосфорилирования. Альтернативным методом фрагментации пептидов, сохраняющим ПТМ в интактном состоянии, является диссоциация с захва-том/переносом электрона, применение которой имеет некоторые особенности, обсуждаемые в представленном обзоре.
ИНСТРУМЕНТАЛЬНЫЕ ОСНОВЫ ТАНДЕМНОЙ МАСС-СПЕКТРОМЕТРИИ
Первые спектры аминокислот, полученные группой Бимана (Biemann) в 1958 г., подтвердили возможность использования методов масс-спек- трометрии для анализа простых пептидов и аминокислот [5]. Дальнейшее развитие масс-спектрометрии пептидов пошло по пути внедрения новых на тот момент методов ионизации — химической ионизации (ХИ, CI), полевой десорбции (ПД, PD), полевой ионизации (ПИ, FI) и электронного удара (ЭУ, EI) в комбинации с газовой хроматографией (ГХ, GC) [6].
Источники ионов с ЭУ давали богатые фраг-ментные спектры, позволяющие получить информацию о структуре вещества, сравнивая информацию с библиотечными спектрами органических соединений. Данный метод позволял получать информацию вплоть до расшифровки отдельных последовательностей белков, подвергнутых гидролизу и переводу в летучее состояние с помощью химической модификации [7]. Источники ионов с ХИ позволяли получать пики протонированных молекулярных ионов аминокислот, проводя их уверенную идентификацию [8], а фрагментация носит индивидуальный характер и зависит от структуры самой аминокислоты и температуры источника. Возможности методов ХИ сильно ухудшаются при переходе к полипептидам, поскольку те отличаются высокой термолабильностью и различные виды дериватизации помогают процессу идентификации только простых цепочек аминокислот [9].
Серьезный прорыв в анализе пептидов и белков был достигнут только с разработкой методов мягкой ионизации. Исторически одним из первых методов мягкой ионизации стал метод плазменной десорбции (ПД) [10]. Этот метод позволял проводить уверенную идентификацию одиночных белков при получении отрицательных молекулярных ионов и даже отдельных нуклеотидов. Делались и множественные попытки адаптировать метод вторично-ионной масс-спектрометрии (ВИМС, Secondary Ion Mass Spectrometry, SIMS) для нужд анализа органических соединений [11]. Однако распространения этот метод не получил вследствие деформации поверхностной кристаллической решетки анализируемого вещества при бомбардировке ионами и уступил место созданному в 80-х гг. более совершенному методу ионизации пептидов бомбардировкой быстрыми атомами (ББА) [12], который фактически стал первым методом, позволявшим определять последовательность аминокислот без дериватизации.
Методы анализа пептидов и белков вышли на новый уровень с развитием действительно мягких методов ионизации, таких как матричная лазерная десорбционная ионизация (МЛДИ, MALDI) [13] и электрораспыление (ЭРИАД, ESI) [14, 15]. Эти методы позволили достигнуть рекордных показателей по чувствительности (уровень аттомолей), работать с гетерогенными образцами, определять белки массой 100 000 Да с погрешностью менее 0.5 % (МЛДИ), а также обеспечить хорошую стыковку с ЖХ, получать многозарядные ионы пептидов и белков весом в миллионы дальтон (ЭРИАД).
Исторически первые тандемные МС-МС приборы пришли из атомной физики, где использовались для исследований точного изотопного состава [16]. Конструктивно они представляли собой каскад из двух анализаторов — магнитного и секторного с ячейкой фрагментации с разреженным газом между ними. Родительские ионы выделялись в магнитном каскаде прибора, а затем, проходя через ячейку фрагментации, анализировались по массе в электростатическом каскаде. Дальнейшее увеличение чувствительности метода МС-МС-анализа (до аттомольного уровня) стало возможно с появлением в 70-х гг. прибора, построенного по системе 3 квадруполей (тройной квадруполь, 3Q) [17], и позднее — ионной ловушки (ion trap, ITMS) [18], включая более позднюю разновидность — линейную ионную ловушку (linear ion trap, LIT) [19]. Данные тандемные приборы позволяли не только получать спектр родительских и дочерних ионов, но и проводить структурные исследования за счет оценки потерь нейтральных частиц при прохождении фрагментации во втором квадруполе. Несмотря на некоторые недостатки (достаточно узкий массовый диапазон, небольшой динамический диапазон, низкие массовую точность и разрешающую способность), тройной квадруполь и ионная ловушка являются одними из самых распространенных приборов для решения задач протеомики.
Для увеличения разрешающей способности и массовой точности был предложен другой тип тандемного МС, в котором в приборе с тройным квадруполем последний квадруполь был заменен на времяпролетный масс-анализатор (ВПА) [20]. Прибор получил название Q-ВПМС (Q-TOF). Такая замена преследовала несколько целей — увеличить скорость записи спектров второй ступенью (до 20 спектров в секунду), повысить разрешающую способность (до 20000–40000) и массовую точность (до 2–3 ppm). Повышение разрешения позволило снизить пороги обнаружения, маскируемые химическим фоном, и улучшить скорость и достоверность идентификации в несколько раз. Замена первой ступени на ВПА-анализатор привела к созданию тандемного прибора ВПА-ВПА (TOF-TOF) с МЛДИ-источником [21]. Этот тип тандемного МС характеризуется умеренной чувствительностью (до одного фемтомоля) и невысокой селекцией в первой ступени (порядка 100), однако хорошо совместим с источниками типа МЛДИ. Дальнейшее развитие TOF-TOF приборов привело к созданию тандемного МС из двух ВПА [22, 23], в котором реализован принципиально но- вый режим "вложенных времен": медленное разделение родительских ионов в первом анализаторе происходит на миллисекундной временнóй шкале, а быстрая фрагментация и быстрый масс-анализ фрагментов во втором анализаторе — на микросе-кундной шкале. Режим вложенных времен позволяет осуществлять так называемый "параллельный анализ" фрагментных спектров для всех родительских ионов в рамках единичного цикла разделения в первом ВПА.
Наиболее высокоразрешающими тандемными приборами в настоящий момент являются МС, использующие преобразование Фурье: ион-циклотронного резонанса ИЦР (ICR FTMS) [24] и масс-анализатора с орбитальным удержанием ионов в электростатическом поле коаксиальных электродов, или "Орбитрэп" (Orbitrap) [25]. ИЦР-анализатор позволяет достигать рекордного значения разрешающей способности, порядка 1000 000 и более, и предела обнаружения менее 1 аттомоль. В тандемном МС с анализатором "Орбитрэп" можно получить разрешающую способность порядка 100000 с высокой массовой точностью порядка 1–2 ppm. В обоих МС ловушка используется для выделения, фрагментации, получения спектра родительских ионов и удаления наиболее интенсивных пиков в спектре, а анализатор — для получения спектра дочерних ионов высокого разрешения (100 000–500 000) с высокой массовой точностью порядка 1 ppm. Вместе с тем к недостаткам приборов данного класса можно отнести высокую стоимость, большие габариты (особенно ИЦР-анализаторы) и длительное время записи спектров в полном массовом диапазоне.
На сегодняшний день для анализа сложных смесей пептидов и белков применяется целый ряд тандемных МС с двумя основными типами источников ионов — МЛДИ и электроспрей. В каждом из этих МС возможна реализация нескольких типов фрагментации первичных ионов в зависимости от выбранной стратегии анализа.
ОСНОВНЫЕ МЕТОДЫ ФРАГМЕНТАЦИИ ИОНОВ В ТАНДЕМНЫХ МС
Индуцированная поверхностью диссоциация (SID)
В методе индуцированной поверхностной диссоциации (surface-induced dissociation, SID) фрагментация происходит при столкновение пучка родительских ионов с поверхностью, при которой происходит образование структурно значимых фрагментных ионов [26, 27]. Степень фрагментации в данном методе может легко регулироваться энергией подаваемого пучка родительских ионов (10–200 эВ). Особенностью метода является высо- кая эффективность конвертирования кинетической энергии во внутреннюю, что позволяет проводить исследования механизмов фрагментации ионов благодаря точному контролю всех параметров эксперимента [28]. Однако в рутинной работе приборы, оснащенные SID-ячейками, применения не нашли. Отчасти это вызвано частичной нейтрализацией родительских ионов при столкновении с фрагментирующей поверхностью, а также образованием заряженных фрагментов поверхности [29].
Фрагментация в источнике ионов (in-source СID)
Фрагментация в источнике ионов (in-source collision-induced dissociation, isCID) — вариант низкоэнергетической фрагментации в источнике ионов "электроспрей", основанный на передаче дополнительной энергии фрагментируемым ионам при их переходе из области атмосферного давления в область низкого вакуума [30]. Установлено, что при повышении напряженности электрического поля в области интерфейса ввода образца в МС увеличивается степень фрагментации исследуемых ионов [31]. Регулировать степень диссоциации можно, изменяя разницу потенциалов между капилляром и скиммером (отбирающим электродом), а в случае улучшенной системы скиммера — между соплом и скиммером [32]. С помощью данного вида фрагментации были получены фраг-ментные масс-спектры пестицидов, лекарственных соединений и пептидов [33]. Дальнейшие работы показали, что дочерние ионы, образующиеся в данном методе, для конкретных соединений аналогичны получаемым в тандемных МС c ячейками фрагментации на основе столкновительной диссоциации (collision-induced dissociation, CID) [34]. Достоинством данного метода является возможность получения фрагментных ионов внутри источника без дополнительной ячейки фрагментации, что позволяет применять его и на обычных, не тандемных МС.
Фрагментация в источнике ионов была применена при анализе сложных биологических образцов, согласно принципам "Шот ган протеомики" (Shotgun Proteomics) с применением длительного ВЭЖХ-разделения [35]. Данный метод реализуем во всех МС, оборудованных источниками ионов "электроспрей", однако широкого распространения не получил.
Столкновительная диссоциация (CID)
Метод фрагментации пептидов "столкнови-тельная диссоциация" (collision-induced dissociation, CID) на данный момент является наиболее распространенным в тандемной МС [36]. В данном методе протонированные пептиды ускоряют- ся электромагнитным полем в вакуумной области МС, а затем подвергаются столкновению с молекулами инертного газа (обычно гелием, азотом или аргоном). Кинетическая энергия пептидного иона частично конвертируется во внутреннюю посредством столкновений. Избыток внутренней энергии перераспределяется в пептидной молекуле, ослабляя и разрывая внутримолекулярные связи, в результате чего образуются фрагментные ионы и нейтральные частицы. Фрагментация пептидов обычно происходит по пептидным связям с получением набора b- и y-ионов, несущего информацию об аминокислотной последовательности пептида [4, 37]. Окончательный спектр фраг-ментных ионов зависит от аминокислотной последовательности и зарядового состояния родительского пептида, времени фрагментации и детектирования. На сегодняшний день наиболее полной моделью диссоциации протонированных пептидов является механизм "направляемой зарядовой фрагментации" [38]. При повышении внутренней энергии молекулы протон становится "мобильным" и может заселять энергетически менее выгодные сайты протонирования (концевая аминогруппа, азот и кислород амидных групп, группы боковых радикалов аминокислот) [39]. Протонирование азотного атома амидогруппы ослабляет пептидную связь, что приводит к ее расщеплению при достаточном уровне внутренней энергии. Т. к. одновременно существует несколько протонированных вариантов одного пептида, образуется набор фрагментных ионов, несущий информацию об аминокислотной последовательности пептида. В общем случае распределение сайтов фрагментации не происходит случайным образом, т. к. боковые радикалы некоторых аминокислот оказывают влияние на локализацию протона [38, 40]. Также энергетически более выгодными могут быть альтернативные пути фрагментации с образованием а- и b-ионов и нейтральными потерями.
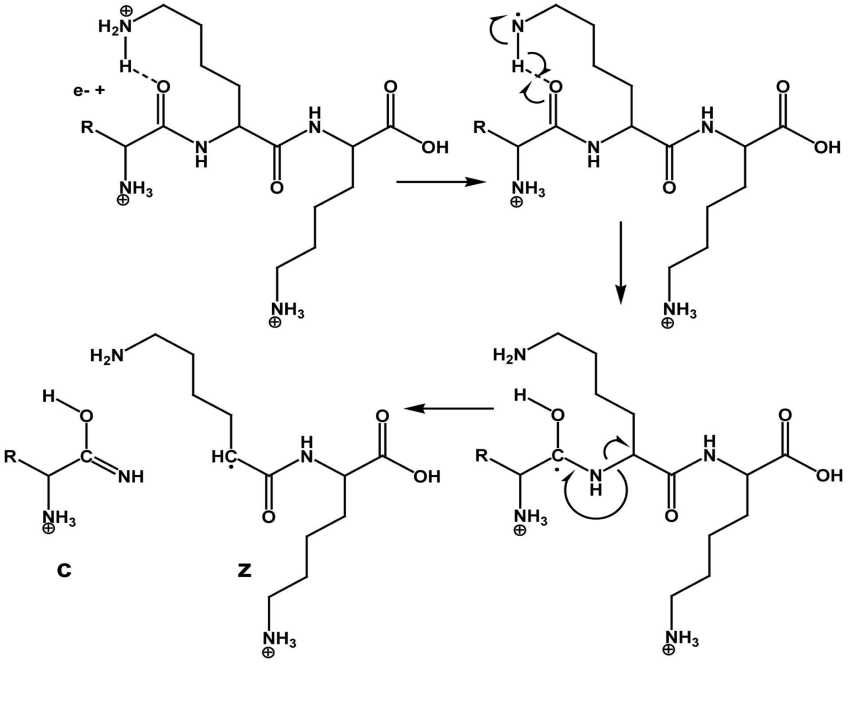
Рис. 1. Предполагаемый механизм ECD- (ETD-) фрагментации.
Присоединение электрона к основному остатку, затем перенос водородного радикала на атом кислорода амидной группы [44]
Фрагментация с захватом электрона (ECD)
Альтернативным методом фрагментации пептидов является диссоциация с захватом электрона (electron-capture dissociation, ECD), предложенная Зубаревым с коллегами в 1998 году. Суть метода заключается во взаимодействии родительского пептида с низкоэнергетическими "тепловыми" электронами, например, в ячейке ИЦР МС, с последующей "мягкой" фрагментацией без передачи пептиду колебательной энергии, как это происходит в случае с методом CID. Однако данный метод фрагментации не может быть использован в МС, использующих радиочастотный захват ионов, из-за кинетического влияния переменного электрического поля на тепловые электроны [41]. Также для получения качественных масс-спектров необходимо усреднение большого количества единичных спектров за большой промежуток времени, что не позволяет использовать ECD в сочетании с ЖХ для анализа сложных смесей.
Диссоциация с переносом электрона (ETD) и ее особенности
В дальнейшем метод ECD был модифицирован [42] и получил название диссоциации при переносе электрона (electron-transfer dissociation, ETD). В таком варианте метод может быть использован в целом ряде тандемных МС, например в МС с радиочастотным квадруполем в качестве ионной ловушки, которые сравнительно недороги, широкодоступны и не требуют больших затрат в обслуживании. Также такие МС позволяют проводить исследования ЖХ-МС в режиме "он-лайн".
В методе ETD протонированные пептиды фрагментируются вследствие переноса электрона (ПЭ) от анион-радикала (например, флуорантена) [42]. В отличие от метода CID в методе ETD происходит разрыв N–Cα связей с образованием серий c- и z-фрагментных ионов (рис. 1). По сравнению с CID в ETD посттрансляционные модификации остаются интактными; таким образом, сохраняется вся информация, необходимая для надежной идентификации пептида [43].
Метод ETD позволяет фрагментировать длинные (более 20 а.к.) многозарядные пептидные ионы, что дает возможность использовать в качестве лизирующих ферментов эндопептидазы Lys-C и Arg-C. В случае использования метода ионизации "электроспрей" полученные пептиды имеют заряд 3–6, что делает их идеальными кандидатами для ETD-фрагментации.
ОСОБЕННОСТИ МЕХАНИЗМА МЕТОДА ДИССОЦИАЦИИ С ПЕРЕНОСОМ ЭЛЕКТРОНА (ETD)
В ион-ионных реакциях перенос электрона от аниона к катиону всегда конкурирует с переносом протона от катиона к аниону [45]. Таким образом, ключевым моментом в ETD является подбор и получение анион-радикалов с минимальным сродством к электрону. По результатам проведенных на сегодняшний день исследований [46] предпочтительным реагентом для ETD является флуорантен, несмотря на то что эффективность переноса электрона для него составляет ~40 %. Таким образом, поиск соединений, обладающих низким сродством к электрону и максимально простой структурой (плоской в идеале), является актуальной задачей в области ETD.
В первых коммерческих МС с ETD в качестве реакционной ячейки и масс-анализатора использовалась линейная квадрупольная ионная ловушка, а в качестве источника анион-радикалов использовалась химическая ионизация (ХИ). Ввод анион-радикалов производился с конца ловушки, противоположного входу положительно заряженных пептидных ионов (метод "back-end") (рис. 2).
Линейная ионная ловушка
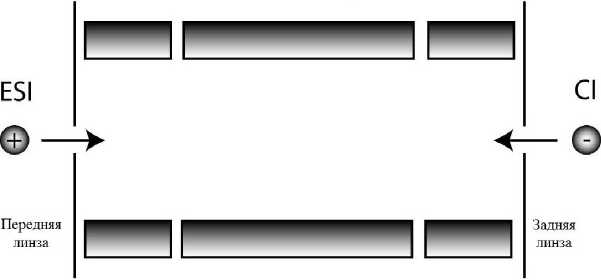
Рис. 2. Схема ввода анион-радикалов в линейную ионную ловушку (метод "back-end") [42]
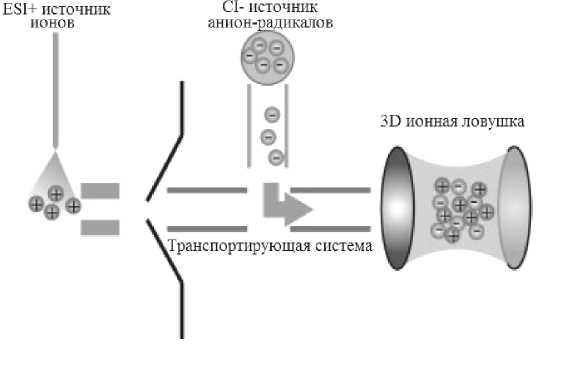
Рис. 3. Схема МС с ETD-фрагментацией и вводом анион-радикалов непосредственно перед ионной ловушкой (в реализации Bruker Daltonics )
В данном варианте метода на получение единичного фрагментного спектра затрачивалось сравнительно малое время (~300 мс), что сделало реальным ЖХ-МС-МС-анализ с высокой производительностью. Однако "задний" ввод анион-радикалов не позволял использовать гибридные варианты распространенных МС (LTQ-Orbitrap, Q-TOF) с дополнительным анализатором высокого разрешения. В обход этого ограничения были разработаны импульсные методы ионизации с последующей активацией реагента-предшественника в CID-ячейке [47]. Так, в методе "двойной импульсный электроспрей" использовалась пара электро-спрей-эмиттеров, один из которых подавал раствор образца, а второй — реагент-предшественник анион-радикалов. Для получения анион-радика-лов в таком варианте метода затрачивалось около 800 мс (200 мс на ввод анионов и по 300 мс на каждое переключение полярности) [48]. Несмотря на это, подобные системы позволили получить фраг-ментные ETD-масс-спектры высокого разрешения (более 60000) и высокой массовой точности (2 ppm). В дальнейшем усовершенствование метода ионизации "двойной электроспрей" привело к уменьшению времени переключения поляризации до 30 мс [49]. Также был предложен метод ввода анион-радикалов в ионную ловушку в средней части ионного тракта непосредственно перед ловушкой, что привело к минимизации временн ы х затрат на получение единичного фрагментного масс-спектра (рис. 3).
Особенности оптимизации механизма ECD
В общем виде механизм фрагментации ETD может быть разделен на два основных канала реакции: перенос протона (proton transfer, PT) и перенос электрона (electron transfer, ET). Перенос PT от мультипротонированных образцов к одноза- рядным анионам проходит легко и является конкурентным процессом по отношению к переносу ET [50]. Продукты PT могут быть обнаружены в виде ионов с редуцированным зарядом, т. к. их фрагментация затруднена. Для получения фраг-ментных ионов таких предшественников с редуцированным зарядом был предложен дополнительной этап активации с помощью модифицированного метода CID (charge-reduced species CID, CRCID). В результате такого дополнительного воздействия образуется фрагментный паттерн, характерный для ETD [51].
Низкую эффективность ETD часто связывают с нековалентными ион-ионными взаимодействиями в ионной ловушке. Дополнительное столкнови-тельное воздействие на такие ионные комплексы вызывает образование непарноэлектронных с- и парноэлектронных z-ионов [52]. Однако сдвиг на 1 Да, вызванный перераспределением атома водорода с с- на z-ион, усложняет анализ получаемых фрагментных масс-спектров [53]. В качестве альтернативного подхода был предложен метод активированных ионов (activated ions, AI) применительно к фрагментации ETD, в котором увеличение внутренней энергии пептидных ионов происходит за счет облучения инфракрасными фотонами [54]. Данный подход похож на ранее разработанную технологию активированных ионов AI ECD, где подобным методом была увеличена эффективность ECD-фрагментации за счет повышения внутренней энергии и, как следствие, ослабления вторичной структуры пептидов [55]. Метод AI ETD резко увеличивает эффективность фрагментации пептидов, благодаря чему количество идентифицированных пептидов становится сравнимо с суммой удачных идентификаций при раздельном использовании методов CID и ETD [54]. Другим преимуществом AI ETD-метода является отсутствие потребности в дополнительном времени на фрагментацию, т. к. все процессы происхо- дят в одном пространстве в одно время.
Также в качестве воздействия, разрушающего нековалентные ион-ионные взаимодействия в линейной ловушке, может быть использовано повышение температуры [56]. Было показано [57], что в случае коротких пептидов (≤ 7 а.к.) возможно полное восстановление их аминокислотной последовательности по сигналам фрагментных ионов при комнатной температуре. Однако перекрытие последовательностей пептидов среднего (8– 11 а.к.) и большого (≥ 14 а.к.) размеров при такой температуре составляет 46 и 23 % соответственно. При повышении температуры эти показатели увеличиваются до 67 и 63 %.
Побочным эффектом данной технологии является появление продуктов термофрагментации, таких как y- и b-ионы, затрудняющих интерпретацию данных. С другой стороны, при использовании МС с точным определением массы наличие дополнительных y- / b- или c- / z-ионных пар облегчает ручное секвенирование пептидов de novo . Повышенный шум детектора также является недостатком метода, делающим невозможным обработку низкоуровневых сигналов [57].
Алгоритм пробоподготовки при использовании метода фрагментации ETD
CID-анализ пептидов, несущих большой заряд, затруднен вследствие резко возрастающего числа сигналов фрагментных ионов, это также делает невозможным комплементарный CID-ETD-анализ. В ходе реакций переноса протонов, проходящих в столкновительной ячейке, может происходить уменьшение зарядности пептидов, но значение m / z получаемых ионов часто не попадает в массовый диапазон МС [42]. В результате стандартного метода обработки белков трипсином основная часть получаемых пептидов несет заряд +2 или +3, что отменяет данную проблему в CID-анализе. Однако эффективность ETD-фрагментации зависит от зарядового состояния родительского иона и становится приемлемой при заряде +3 и выше. Таким образом, одной из задач, стоявших перед исследователями, была разработка пробоподготовки с получением пептидов с зарядом ≥ +3 [44]. Формально пробоподготовка состоит из нескольких фаз.
-
1 . Использование альтернативных протеаз. Одним из подходов является использование альтернативных протеаз, таких как Lys-C и Asp-N, которые расщепляют белок на бóльшие фрагменты (20–25 а.к.), чем трипсин (10–15 а.к.). С увеличением числа основных аминокислотных остатков в составе пептида увеличивается и их заряд. Также уменьшается количество ложнопозитивных результатов, т. к. с увеличением длины пептида уменьшается количество возможных
-
2. Дериватизация пептидов. С помощью N-гидроксисукциниминового эфира 4-триметил-аммоний бутирата можно дериватизировать первичные амины пептидов. Из-за того что заряды на таких модифицированных пептидах фиксированы, эффективность их фрагментации снижается. Однако при этом сильно увеличивается интенсивность фрагментных ионов, получаемых при расщеплении дисульфидных связей, по сравнению с интактными пептидами, что делает данный метод пригодным для исследования цистиновых мостиков в белках [60].
-
3. Модификации растворителя. В качестве агента, вызывающего увеличение среднего зарядового состояния молекулярных ионов в газовой фазе (генерируемых в электроспрее), может быть использован m-нитробензиловый спирт (NBA) [61]. Способность NBA увеличивать заряд анали-тов связывают с низким давлением пара в сочетании с высоким поверхностным натяжением данного соединения. Эти свойства препятствуют достижению рэлеевского предела, после достижения которого капли, образующиеся в методе ионизации "электроспрей", испытывают кулоновский взрыв. Большая плотность зарядов в таких каплях в итоге трансформируется в большее среднее значение заряда анализируемых соединений.
вариантов, которым может соответствовать данная аминокислотная последовательность (при работе с базами данных).
В последнее время были разработаны два методических подхода в пробоподготовке для ETD МС-анализа. Первый основан на неферментативном расщеплении белков с помощью микроволнового излучения, специфически разрывающего С-терминальную связь остатка аспарагиновой кислоты. Данный метод позволяет получать пептиды размером 15–25 а.к. остатков с зарядом z > +3. Микроволновое расщепление белка происходит в течение нескольких минут (< 6 мин). Получаемые пептиды могут быть успешно фрагментированы с помощью ETD для получения полного набора c- / z-ионов, необходимых для уверенной идентификации. Быстрота и совместимость метода с ETD делают его идеальным инструментом для протеомики [58].
Во втором подходе используется обработка белка протеазой Lys-N с получением двухзарядных пептидов, при этом заряд локализован на N-конце аминокислотной последовательности. Таким образом, одиночный заряд несут только с-ионы, z-ионы нейтральны и не могут быть детектированы в масс-анализаторе. Эффективность ETD-фрагментации Lys-N пептидов достаточно высока и позволяет получать полные наборы с-ионов для de novo секвенирования [59].
Воздействие NBA на зарядовое состояние пептидов также было исследовано в [62]. Отмечено возрастание среднего зарядового состояния триптических пептидов сывороточного альбумина быка (BSA) с +2.2 до +2.6 при добавлении NBA в растворитель до конечной концентрации 0.1 %. Большая часть пептидов BSA в таком варианте электроспрея несла заряд +3. Эффективность фрагментации таких пептидов значительно выше по сравнению с фрагментацией триптических пептидов в обычном растворителе, что увеличивало возможность идентификации по базе данных белков Mascot в среднем на 68 %.
Данный подход может быть легко реализован добавлением 0.1 % NBA в элюенты при хроматографическом разделении пептидов или присоединением Т-образного сплиттера непосредственно перед источником ионов. Другим преимуществом NBA является совместимость с методом триптического гидролиза белков. Стоит, однако, иметь в виду, что молекула NBA имеет гидрофобные свойства и при добавлении в подвижную фазу уменьшает время удерживания пептидов на обращенно-фазовых колонках (линейная зависимость от концентрации NBA в подвижной фазе).
В среднем заряд различных пептидов после обработки NBA увеличивается на 0.25. Триптические фосфопептиды редко несут заряд более +2, их однозначная идентификация и локализация фосфогруппы затруднены в условиях ETD. При добавлении NBA к образцу заряды фосфопептидов возрастают в среднем примерно на 0.5 зарядовой единицы.
ВОЗМОЖНОСТИ АНАЛИЗА ФОСФОРСОДЕРЖАЩИХ ПЕПТИДОВ С ПОМОЩЬЮ ТАНДЕМНЫХ МС
С CID- И ETD-МЕТОДАМИ ФРАГМЕНТАЦИИ
Одним из важнейших преимуществ ETD-технологии в протеомике является возможность локализации точного сайта посттрансляционной модификации (ПТМ) белков, что не всегда доступно в случае CID.
Обратимое фосфорилирование белков является одним из наиболее распространенных механизмов регуляции процессов, происходящих в клетке. Этот механизм регулирует такие биологические процессы, как межклеточная коммуникация, клеточный рост, размножение, дифференциация и апоптоз [63]. Фосфорилирование влияет на основные свойства белков, модулируя фолдинг, активность, белок-белковые взаимодействия, внутриклеточную локализацию и деградацию белка. Низкая стехиометричность фосфопептидов в сочетании с отрицательным зарядом фосфогруппы делает их анализ в режиме регистрации положительных ионов затруднительным. При анализе с помощью CID-фрагментации остаток фосфотирозина довольно стабилен, в то время как остатки фосфосерина и фосфотреонина лабильны, т. е. об их наличии можно судить только по нейтральным потерям, соответствующим массе фосфорной кислоты (сдвиг на 98 Да) [64].
В отличие от CID, ETD-фрагментация позволяет получить практически полные серии c- и z-ионов (рис. 4), делая возможным протеомный анализ фосфопептидов.
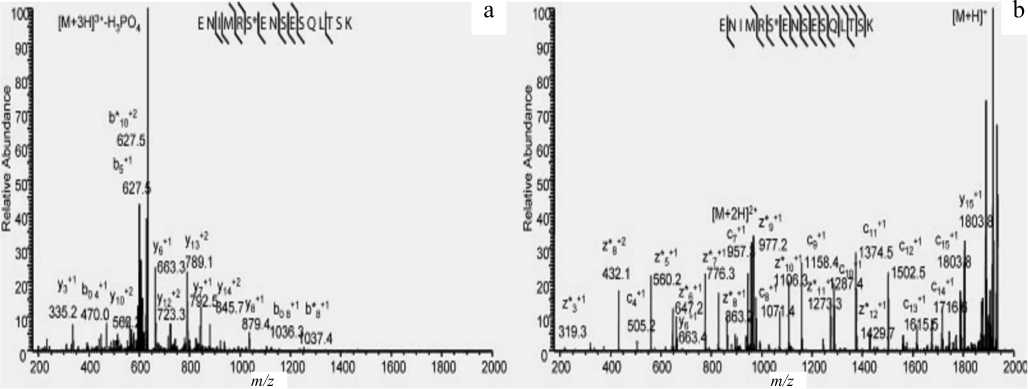
Рис. 4. CID (а) и ETD (b) фрагментные спектры трехзарядного протонированного синтетического пептида ENIMRS*ENSESQLTSK, имеющего фосфатную модификацию ( * ).
Спектры получены с помощью МС LTQ XL ( Thermo Scientific ) [44]
Первые эксперименты с использованием ETD-фрагментации для исследования фосфопептидов проводились на дрожжевых клетках. Исследователи использовали эндопептидазу Lys-C, получая большие и многозарядные пептиды. В результате одного эксперимента с использованием нанопото-ковых систем в 20 мкг клеточного лизата было идентифицировано 1252 сайта фосфорилирования в 629 белках, включая нестабильные остатки фосфогистидина [65].
Недавно группа Молино с сотрудниками идентифицировали 1435 фосфопептидов из клеток 293Т почек эмбриона человека, из них 80 % не были описаны ранее. В данном исследовании было проведено сравнение результатов, полученных с использованием CID и ETD. В случае использования ETD-фрагментации количество идентифицированных фосфопептидов было на 60 % больше, чем при использовании CID [43]. Для расщепления полипептидной цепи применялась эндопептидаза Lys-C, т. к. использование трипсина слегка уменьшало статистику идентификаций. Также было обнаружено, что при использовании трипсина среднее количество пропущенных сайтов гидролиза (1.1) и длина получаемых пептидов (16) в случае фосфопептидов выше, чем в случае нефосфо-рилированных (ср. количество пропущенных сайтов расщепления 0.3, средняя длина пептидов 12).
Множественные сайты фосфорилирования, расположенные рядом в а.к.-последовательности, требуют полного набора серий фрагментных ионов для определения точного сайта модификации. Преимущества сочетания методов CID и ETD проявляются при анализе фосфопептидов, чьи сайты модификации расположены рядом с пролином. ETD-фрагментация не позволяет расщеплять такие химические связи, а CID может быть использована для получения активированных родительских ионов. Недавние исследования с использованием статистической обработки крупномасштабных экспериментов, в которых для фрагментирования элюируемых с нано-ВЭЖХ колонок пептидов поочередно использовались CID и ETD [66], показывают, что при идентификации фосфопептидов, несущих двойные модификации, получаемые данные сильно различаются в зависимости от метода фрагментации и дополняют друг друга. Так, между группами идентифицированных с помощью CID (141) и ETD (79) пептидов наблюдается пересечение лишь в 8 % (17) от общего числа идентифицированных. Стоит отметить, что у пептидов, идентифицированных обоими методами, различия в определении сайта модификации наблюдались только в трех случаях. Причем причиной таких различий было низкое качество фрагментных масс-спектров, обнаруживаемое при анализе "вручную". Также авторы отмечают отсутствие признаков перераспределения фосфогрупп между несколькими возможными сайтами модификации в монофосфорилированных пептидах, что предполагалось ранее для CID-фрагментации [67].
ОСОБЕННОСТИ ИНТЕРПРЕТАЦИИ ДАННЫХ И БАЗЫ ДАННЫХ ФОСФОПЕПТИДОВ ПРИ ИСПОЛЬЗОВАНИИ МЕТОДА ETD
Ранние исследования с применением ETD-фрагментации основывались на ручной обработке МС-МС-спектров с разметкой c- и z-ионов. На данный момент разработано несколько алгоритмов для проведения автоматической интерпретации масс-спектрометрических данных и локализации сайтов фосфорилирования: MASCOT [68], OMSA [69], SEQUEST, Specrum Mill, X!Tandem [70]. Для обработки данных, полученных с использованием метода ETD, наиболее применимыми считаются Spectrum Mill и MASCOT [71]. Protein Prospector также показал эффективность обработки данных после оптимизации системы подсчета [72].
При фрагментации методом ETD пептиды, полученные обработкой белка эндопептидазой Lys-N, в основном образуют фрагментные ионы с-типа [59, 79]. На основе данной особенности был создан алгоритм LysNDeNovo для de novo секвенирования таких пептидов [73]. Для обработки ETD-МС-МС-данных был разработан отдельный алгоритм поиска по существующим базам данных — Z-core. Этот алгоритм показал минимальное преимущество по отношению к OMSA при идентификации пептидов [74]. Основные инструменты и базы данных фосфопротеомов представлены в таблице (см. Приложение).
Хотя на данный момент для метода ETD разработаны алгоритмы поиска в уже существующих базах данных (Z-core, MS-GFDB, pFind 2.1) и секвенирования de novo (CompNovo, LysNDeNovo), есть ряд методов, в которых применимость таких алгоритмов невозможна. Например, в случае использования CID- и ETD-методов фрагментации последовательно в течение одного МС-МС-анализа [75].
ВЫВОДЫ
В данном обзоре изложены возможности и ограничения методов фрагментации фоcфопептидов в тандемной масс-спектрометрии с использованием столкновительной диссоциации CID и диссоциации с переносом электрона ETD. На данный момент масс-спектрометрические методы проте-омного анализа в достаточной мере сформированы для приборов, использующих CID-фрагментацию. В то время как CID-фрагментация хорошо зареко- мендовала себя при работе с короткими и средними пептидами (до 11 а.к.), метод фрагментации ETD показывает наилучшие результаты при анализе длинных (> 14 а.к.) и высокозаряженных (z ≥ +3) пептидов. В этом случае для гидролиза белков рекомендуется использовать альтернативные трипсину протеазы, такие как Lys-C, Asp-N, Lys-N, либо проведение неферментативного гидролиза белков с помощью микроволнового излучения. Результатом таких вариантов гидролиза белков становятся пептиды с длиной аминокислотной цепи 15–25 остатков, что увеличивает количество сайтов протонирования и приводит к повышенному зарядовому числу. В случае электро-спрей-ионизации возможно использование моди-фикантов растворителя, также увеличивающих среднее зарядовое число пептидов гидролизата. Оба метода пробоподготовки обеспечивают эффективную фрагментацию в случае использования ETD. Использование метода фрагментации ETD приводит к получению фрагментных ионов "c" и "z" (в отличие от метода фрагментации CID, дающего b- и y-ионы), что приводит к использованию существенно другого подхода к обработке данных фрагментных масс-спектров. В настоящее время продолжается активная разработка алгоритмов интерпретации фрагментных масс-спектров, полученных с помощью метода ETD, в том числе с использованием существующих электронных баз данных по фосфопротеомике, что видно из представленной таблицы (Приложение).
Проведенный анализ опубликованных данных показывает, что при решении задач протеомики результаты, получаемые с применением методов фрагментации CID и ETD, комплементарны. Кроме того, сохранение в интактном состоянии посттрансляционных модификаций белков и пептидов при использовании метода фрагментации ETD позволяет получить больше информации о состоянии биологических образцов. Таким образом, большим преимуществом тандемных МС, использующих оба метода фрагментации CID / ETD, является значительное увеличение информативности единичного эксперимента, а следовательно, расширение возможностей по восстановлению аминокислотных последовательностей. Стоит ожидать, что в ближайшем будущем ETD станет общепринятым методом тандемной масс-спектрометрии для решения задач протеомики в одном ряду с CID.
Приложение
Доступные протеомные инструменты и базы данных фосфопептидов
Прогнозирование сайтов модифицирования Название URL Описание Ссылка NetPhos services/NetPhos/ Прогнозирование сайтов модифицирования белков эукариот по серину, треонину и тирозину с помощью нейронных сетей [76] NetPhosK services/NetPhosK/ Прогнозирование киназ-зависимого фосфорилирования белков эукариот с помощью нейронных сетей [77] NetPhos Yeast services/NetPhosYeast / Прогнозирование сайтов фосфорилирования по серину и треонину дрожжевых белков [78] Протеомные инструменты Protein Prospector edu/prospector/ Протеомные инструменты для поиска в базах данных белковых последовательностей совпадений с результатами МС-эксперимента [78]
Продолжение таблицы
Локализация и оценка корректности назначения сайтов фосфорилирования Название URL Описание Ссылка APIVASE software/ API- Автоматическая идентификация и валидация SEQUEST. Корректность идентификации проводится сравнением информации, полученной обработкой спектров МС2 и соответствующих спектров нейтральных потерь МС3 [79] Ascore Оценка вероятности корректного определения сайта фосфорилирования по наличию и относительной интенсивности сигналов в МС-МС-спектре [80] SLoMo slomo Расчет гипотетических CID или ETD/ECD фрагментных ионов на основе базы данных ранее определенных модификаций UniMod [81] MSQuant http://msquant. Количественное определение белков на основании интенсивностей родительских ионов (SILAC/ICAT/ICPL); МС3-скоринг идентификации пептидов; ПТМ-скоринг вероятности локализации сайта модификации [82] ProPhosSI http://www.compbio. prophossi/bin/ Валидация совпадения экспериментального спектра фосфопептида с имеющимися в базе данных [83] Базы данных P3DB База данных фосфорилированных белков и пептидов растений (Plant Protein Phosphorylation Database) [84] Peptide Atlas http://www. Публично доступная база данных пептидов, обнаруженных МС-МС-методами в различных организмах [85] Phosida http://www.phosida. com/ Каталогизация, структурные и эволюционные исследования, прогнозирование сайтов фосфорилирования; данные по фосфорилированию, ацетилированию и N-гликозилированию белков [86] Phospho ELM org/ База данных экспериментально подтвержденных S/T/Y сайтов фосфорилирования эукариот [87] PhosphoGRID http://www. База данных экспериментально подтвержденных сайтов фосфорилирования почкующихся дрожжей Saccharomyces cerevisiae [88]
Продолжение таблицы
Название URL Описание Ссылка PhosphoNET http://www. База данных по сайтам фосфорилирования человека; оценка эволюционной консервативности; теоретический подбор возможных киназ, действующих по конкретному сайту PhosphoPep http://www. Проект по поддержке системных исследований биологического сигналинга. Предоставляются данные, полученные МС-методом, по объектам: Drosophila melanogaster, Homosapiens, Caenorhab-ditiselegans, Saccharomyces cerevisiae [89] PhosPhAt http://phosphat. База данных по сайтам фосфорилирования Arabidopsis [90] PhosphoPoint http://kinase. База данных по киназам и фосфопептидам человека [91] PhosphoSitePlus http://www. Фосфопротеомика и прочие ПТМ [92] RESID Database RESID; /RESID Сборник аннотаций и структур и модификаций (пре-, ко- и посттрансляционных) пептидов [93]
