Hybrid Machine Learning Approaches for DNA Classification: A Stacking Classifier Perspective

Автор: Sultanul A. Hamim, Dip Nandi, Niloy E. Costa
Журнал: International Journal of Intelligent Systems and Applications @ijisa
Статья в выпуске: 3 vol.18, 2026 года.
Бесплатный доступ
This paper presents a hybrid machine learning model for the classification of DNA sequences by combining different machine learning algorithms, including K-Nearest Neighbors (KNN), Support Vector Classifier (SVC), Decision Tree, Random Forest, Light Gradient Boosting Machine (LGBM), and XGBoost (XGB). This model has been developed using the stacking ensemble method, associated with a majority voting mechanism to achieve improved overall classification accuracy. In this study, the Promoter Gene Sequences dataset from the UCI Machine Learning Repository was used to concentrate on classifying promoter versus non-promoter sequences. The results indicated an accuracy of 96.25%, showcasing the hybrid model’s ability to classify DNA sequences effectively. This research provides valuable insights into ensemble machine-learning techniques in DNA classification, with possible applications in genomics research, medical diagnostics, agricultural biotechnology, and forensic science. The hybrid model’s thriving implementation demonstrates the potential for more accurate and reliable DNA sequence classification methods.
DNA Sequence Classification, Machine Learning, Hybrid Models, Bio-informatics, Predictive Analytics
Короткий адрес: https://sciup.org/15020397
IDR: 15020397 | DOI: 10.5815/ijisa.2026.03.07
Текст научной статьи Hybrid Machine Learning Approaches for DNA Classification: A Stacking Classifier Perspective
Published Online on June 8, 2026 by MECS Press
Deoxyribonucleic acid (DNA) is the primary hereditary material in humans and nearly all other organisms, playing a crucial role in the development, growth, and reproduction of all known life forms and many viruses. It has a typical double-stranded structure with sugar-phosphate backbones wrapped around a pair of nitrogenous bases [1]. This molecular blueprint, housed mainly in the cell nuclei, contains genetic information used in identifying species, studying evolution processes, and investigating the functional genomics area [2]. It was just the invention of new sequencing technologies, which started representing databases with oceans of genetic data in terms of billions of base pairs, thereby marking the tremendous genomics information growth [3]. DNA sequence classification is a very pertinent problem in genomics with far-reaching implications on several domains, such as biomedicine, agriculture, and forensic science [4]. Computational methods are becoming an essential tool for management and interpretation of these large datasets [5]. In the analysis and classification of DNA sequences, important tools have proved to be the following machine learning algorithms: Nearest Neighbors, Gaussian Process Classification, Decision Trees, Random Forests, Neural Networks, AdaBoost, and Support Vector Machines [6]. Each of them has its own special advantage: KNN has equality in similarity-based classification, GPC makes probabilistic predictions that are important in confidence assessment, Decision Trees provide transparent structures of decisions to identify genetic markers, Random Forests ensure robustness against data variability, Neural Networks are able to grasp some intrinsic sophisticated structures of the genomic data, AdaBoost combines weak learners while boosting the classification accuracy, whereas SVM maximizes margins of classes in separable datasets, hence
This work is open access and licensed under the Creative Commons CC BY 4.0 License.
optimizing high-dimensional DNA analysis [7, 8]. Although these Machine-Learning models have immense power, each of them has intrinsic limitations, making them use judiciously [9]. Computation time with SVM increases as a function of the size of the dataset, and Decision Trees can overfit genetic data. Random Forests avoid overfitting by averaging many decisions trees, however at the cost of lost interpretability. The example highlights that appropriate choice among ML techniques shall answer specific genomic datasets and analytical objectives [10, 11]. The presented work deals with the integration of a 6 ML models into a hybrid approach for DNA sequence classification with accuracy above 96.25%. This paper tries to solve problems associated with accuracy, robustness, and computational efficiency traditional techniques face by revealing the strengths of their diverse algorithms. Not only do these findings help promote further genomic research, but the result also shows some potential for increasing precision in medicine and agricultural biotechnology with forensic genetics applications.
2. Literature Review
The application of machine learning to biological data analysis has revolutionized our ability to interpret complex genetic and genomic information. By leveraging various machine learning models, researchers can classify and predict outcomes in DNA sequencing and gene expression with unprecedented accuracy. This section delves into recent advancements and methodologies in machine learning for DNA sequence and gene expression data, highlighting key techniques and their performance in tackling real-world biological challenges.
-
2.1. Machine Learning
-
2.2. Deep Learning Models
-
2.3. Hybrid and Novel Approaches
-
2.4. Logistic Regression and Ensemble Approaches
Machine learning plays a pivotal role in genetic and genomic studies, offering diverse models for complex classification tasks. Recent studies have demonstrated the effectiveness of both traditional machine-learning techniques and advanced deep learning models for DNA sequence classification. For example, the Modified k-Nearest Neighbour (MKNN) technique, including scenarios such as SMKNN and LMKNN, has shown superior performance in handling high-dimensional gene expression data with reduced testing times, outperforming traditional models like KNN, weighted KNN, SVM, and brain emotional learning (BEL) in terms of accuracy, precision, and recall [12].
Methods such as SVM, CNN, Random Forest, and XGB Classifier have also been applied to DNA sequencing, with XGB achieving the best performance at 92.2%, followed by SVM at 89.4%, while Adaboost underperformed at 42.2% [6]. Deep learning approaches further enhance classification tasks; for instance, a neural network leveraging spectral sequence representation achieved higher accuracy than naive Bayes 86%, random forest 85%, and SVM 84%, particularly for fragments of 500 base pairs [13]. Similarly, integrating CNN with tree-based methods, such as random forest and gradient-boosted trees, resulted in high-performing models, with CNN-generated scores boosting the accuracy of gradient- boosted trees and random forest to 89.6%, compared to SVM at 89.0% [14]. These advancements underscore the growing potential of machine learning and deep learning in decoding complex genetic information, offering increasingly accurate and efficient solutions for DNA sequence classification [15].
Recent advances in deep learning have yielded a dramatic increase in the performance of DNA sequence classification. Deep learning neural networks, particularly convolutional neural networks, have shown good performance compared to traditional classifiers. A Deep learning framework using spectral representation of the sequence outperformed Naive Bayes- 86%, random forest (85%), and support vector machines 84% in classifying small DNA sequence fragments of 500 base pairs [13]. Other developments included CNNs combined with tree-based methods, which illustrated that CNN- generated score features combined with features from random forests and gradient boosted trees reached an accuracy of 0.896, surpassing SVM’s 0.890 [14]. he CNN and CNN-Bidirectional LSTM models with K-mer encoding had the impressive accuracy rates of 93.16% and 93%, respectively, with suitability of these models for high-dimensional data demonstrating their application in virus detection and drug design [16].
Advanced in hybrid and novel methods amped the classification performance on complex DNA sequences. DBPPred integrates feature ranking with random forest and wrapper-based selection, utilizing Gaussian Naive Bayes to predict DNA-binding proteins. This method achieved an accuracy of 0.791 and an MCC of 0.583 on the PDB594 dataset, outperforming other models [17]. Meanwhile, parallel to this, some new techniques are being developed for the helitron DNA sequence classification in C. elegans use Frequency Chaos Game Representation combined with machine learning approaches. This framework, with FCGR images and K-mer features, reached the highest global classification accuracy of 72.7% using various classifiers [18]). These contributions show that a possible benefit derived from the combination of different approaches for an increase in accuracy with respect to prediction and efficiency in classification at genomic level.
Table 1. Summary of machine learning model performance for DNA and genetic sequence classification
|
Model |
Accuracy |
Reference |
|
DBPPred |
0.769–0.791 |
[17] |
|
Random Forest + CNN |
0.896 |
[14] |
|
Gradient Boosting Machines |
89.6% |
[14] |
|
MKNN |
70-90% |
[12] |
|
CNN and Random Forest |
92.2% |
[6] |
|
Deep Learning Neural Network |
86% |
[13] |
|
CNN |
93.16% |
[16] |
|
CNN-Bidirectional LSTM |
93.13% |
[16] |
|
Naive Bayes |
83.8% |
[15] |
|
Various ML Techniques |
Over 90% |
[21] |
|
FCGR |
72.7% |
[18] |
|
Random Forest |
92.2% |
[6] |
|
AdaBoost |
42.2% |
[6] |
|
CNN |
92.2% |
[6] |
|
k-NN |
70-90% |
[12] |
|
Logistic Regression |
85-95% |
[19] |
|
Decision Trees |
90% |
[21] |
|
Naive Bayes |
83.8% |
[15] |
Recent advancements in logistic regression and ensemble approaches have significantly improved genetic sequence classification and DNA microarray analysis. A machine learning logistic regression pipeline was developed to handle genetic data efficiently, achieving high accuracy 85-95% even with incomplete data and near-perfect accuracy with com- plete datasets, while also identifying key amino acid positions for clade classification. Meanwhile, optimizing ensemble classifiers through correlation-based feature selection techniques has enhanced performance in DNA microarray classification by addressing mutual errors among classifiers. This method has demonstrated superior recognition rates across three benchmark datasets, effectively combining diverse feature sets [19, 20].
This table 1 presents an overview of various machine learning models used for DNA and genetic sequence classification, highlighting their accuracy and sources. It includes traditional classifiers like Random Forest, k-NN, and Naive Bayes, as well as advanced methods such as deep learning neural networks and ensemble techniques. The table showcases the effectiveness of different models across diverse datasets and applications, illustrating their potential for improving classification accuracy in genomics and bioinformatics.
3. Methodology
Accurate classification of DNA sequences is of prime importance in several biological and medical applications. Herein, we have projected a hybrid machine learning model to classify promoter sequences and nonpromoter sequences with high accuracy and provide an outlook on how it may become much better at improving predictive performance and reliability while carrying out DNA sequence classification tasks by benefiting from multiple algorithms. The next parts describe the methodology and performance evaluation of the proposed hybrid model for classifying promoter and nonpromoter sequences.
-
3.1. Data Collection and Data Preprocessing
The dataset used in this study was the Promoter Gene Sequences dataset, available from the UCI Machine Learning Repository. The dataset contained DNA nucleotide sequences, each tagged as a promoter or non-promoter. This dataset contained 106 instances, each 57 long; the class associated showed whether a test sequence was a promoter or not a promoter. Figure 1 show the sample image of the dataset.
Data preprocessing can be very instrumental in rendering data into some format to make it fit for use by the machine learning algorithm. Several steps have been taken in the current study to ensure that this dataset is transformed into a form suitable for model training and evaluation. Such will include loading the dataset, transforming the nucleotide sequences, encoding categorical variables, feature scaling, and splitting datasets into train and test datasets. Numerical encoding was applied to nucleotide sequences and class labels to make the data compatible with machine
Sample of the Dataset:
0123456789 Otactagcaat Itgctatcctg 2gtactagaga 3aattgtgatg 4tcgataatta
48 49 50 51 52 53 54 55 56 Class gcttgtcgt+ catcgccaa+ cacccggcg+ aacaaactc+ ccgtggtag+
Fig.1. Sample Image of dataset learning algorithms. LabelEncoding was selected for this purpose because it assigns a unique integer to each nucleotide (A, T, C, G), preserving the categorical nature of the data while maintaining computational efficiency. Compared to other encoding techniques, such as one-hot encoding, LabelEncoding avoids introducing unnecessary dimensionality, which is particularly beneficial for small datasets like this one. For class labels, encoding was done similarly, where the class of promoter sequences was set to 1, while non-promoters were set to 0.
To enhance the performance of the model, feature scaling was applied using the StandardScaler. StandardScaler standardizes features by removing the mean and scaling to unit variance, ensuring that all features contribute equally during model training. Additional steps were taken to ensure the data preparation accounted for potential variations. Synthetic noise was introduced during preprocessing to evaluate the model’s performance under less ideal conditions. The dataset’s class distribution was also analyzed and confirmed to be balanced, helping mitigate concerns about data imbalance. Although the dataset inherently consists of sequences of uniform length (57 nucleotides), preprocessing ensured that variations in sequence length, if present, would not affect the model’s performance. Finally, the dataset was divided into a training set (75%) and a test set (25%) for model evaluation.
-
3.2. Model Development
The first step in this study was to build and assess thirteen diverse individual machine learning models. These models were selected to represent a wide range of machine learning techniques, enabling a thorough evaluation of different methodologies for classifying DNA sequences. Specifically, the models deployed were Logistic Regression, K-Nearest Neighbors, Decision Tree, Random Forest, Support Vector Machine, Naive Bayes, Gradient Boosting, AdaBoost, Nearest Centroids, Bagging Classifier, Multi-Layer Perceptron, Perceptron, and LightGBM.
Each model was trained on the training set, which comprised 75% of the entire dataset. To optimize their performance, key hyperparameters were tuned using systematic approaches such as GridSearchCV and RandomizedSearchCV. Random Forest hyperparameters included the number of estimators (n estimators), maximum depth (max depth), and minimum s a mples spli t (min samples split), with ranges of 50–200, 5–20, and 2–10, respectively. Light- GBM tuning focused on the learning rate (learning rate), maximum depth (max depth), and number of leaves (num leaves), with ranges of 0.01–0.2, 3–10, and 20–50, respectively. Gradient Boosting tuning included the learning rate (learning rate), number of estimators (n estimators), and subsample ratio (subsample), with ranges of 0.01–0.1, 100– 300, and 0.6–1.0, respectively. Simpler models, such as Logistic Regression and Naive Bayes, used default configurations where applicable, ensuring fairness in evaluation.
Hyperparameter tuning was performed with five-fold cross-validation to prevent overfitting and identify the best con- figurations for each model. This systematic approach ensured that all models were optimized for the classification task while maintaining robustness. After training, the models were evaluated using the remaining 25% of the data, referred to as the testing set. Metrics such as accuracy, precision, recall, and F1-score were used to provide a detailed perspective of each model’s performance. This comprehensive evaluation enabled the selection of high-performing base models for integration into the hybrid framework.
-
A. Hybrid Model Construction
The construction of the hybrid model was to integrate all the strengths existing in the different base models that perform best in DNA sequence classification for a better prediction performance. In the overall creation of this model, a few major steps had to be considered.
First, the top six base models based on their respective evaluation metrics of accuracy, precision, recall, and F1 -score were selected. The selected models at this point were Random Forest, LightGBM, Gradient Boosting, AdaBoost, SVM, and MLP. All these selected models were individually trained on the training dataset to ensure the model was well-optimized for making suitable predictions on DNA sequences. Afterwards, the models were use from the testing dataset. The predictions thus obtained were then used as input features for constructing the hybrid model. That is, for every instance from the test set, the output of each base model represented a new feature set in which every instance is represented by the output of the six base models. Mathematically this can be written as:
P svc (y = 1 | X),P DT (y = 1 I X),TMy = 1 I X),? LGBM (y = 1 I X),P xGB (y = 1 Ш (1)
/ ^ KNN (У — 1 1 ^) \ ' Psvc (у —mo ' P DT (y — H^) P rf (y —im
^ LGBM (y — 1 1 ^ ) , \ ^ XGB (У — 1 | X)7
For these predictions a stacking ensemble method was used. First, metafeatures were created out of the combinatorial prediction factors of the base learners. The major meta-model in use is the logistic regression model that learned in the best way how to come up with the final classification based on combining the individual predictions. The meta-model was made to operate on the combinational predictions of Z and computes a linear combination with weights W stack and bias b stack , producing the final prediction score Z stack .
T
Z stack W stack Z + b stack
The sigmoid function σ ( Z stack ) transforms this score into a probability value between 0 and 1, representing the likelihood of the positive class:
P stack ( У = 11 Z ) = <4 Z stack ) = ^
1 + e z stack
In addition to the mentioned stacking method, a majority voting mechanism was added to determine the final class prediction for each instance. Here, each of the six base models voted for its predicted class, and the class with many votes was chosen as the final prediction. This is yet another way that the hybrid model enhances its robustness and accuracy through a voting mechanism, in which one can obtain a consensus from multiple algorithms. Final Prediction y final is thus determined by:
y^, = Mode{yM 1 , yM 2 ,..., yM 6 , y mea } (4)
The architecture of this hybrid model employs the base models to be fine-tuned, and after training the selected base models, they generate predictions used for the creation of a new feature set, while the meta-models train to amalgamate them. By combining the strengths of the diverse nature of machine learning algorithms, the hybrid model here achieves an accuracy above 96.2% in classification. The model worked very well in classifying DNA sequences. This tendency emphasizes high performance in the hybrid model as a predictor for various complex classification cases.
The hybrid model leverages stacking ensemble and majority voting techniques, which are powerful methodologies in machine learning, to integrate the strengths of diverse algorithms. While these methods are established, their strategic application in DNA sequence classification addresses a critical gap in genomics research. The choice of these techniques was driven by their ability to combine the predictive power of traditional models such as Random Forest and SVM with advanced algorithms like LightGBM and XGBoost, resulting in a balanced and robust approach. This hybrid framework provides a scalable solution to handling high-dimensional and noisy datasets, which are common challenges in DNA sequence analysis, demonstrating the utility of ensemble techniques in this specific domain.
The hybrid model leverages stacking ensemble and majority voting techniques, which are powerful methodologies in machine learning, to integrate the strengths of diverse algorithms. While these methods are established, their strategic application in DNA sequence classification addresses a critical gap in genomics research. The choice of these techniques was driven by their ability to combine the predictive power of traditional models such as Random Forest and SVM with advanced algorithms like LightGBM and XGBoost, resulting in a balanced and robust approach. This hybrid framework provides a scalable solution to handling high-dimensional and noisy datasets, which are common challenges in DNA sequence analysis, demonstrating the utility of ensemble techniques in this specific domain.
The evaluation of the proposed hybrid model should give a view regarding its classification effectiveness in DNA sequence, not only of the vis-a`-vis based-model approach. This section will present detailed results, hence including performance through confusion matrix measures, ROC curve analysis, and comparative benchmarking with the based models, reflecting how the hybrid approach is solid and reliable. The classification of DNA sequences was performed with the individual base models and tested for their accuracy, precision, recall, and F1 score. The results of the performance metrics of individual base models are shown. Regarding individual base models for performance based on accuracy, the best performers were Random Forest and LightGBM models, with accuracy scores of 92.59% and 93.30%. It has been done that except for the mentioned models, quite high precision, recall, and F1-scores were also in the other, which showed that the feature vectors used are able to predict the class of positive and negative classes with much proficiency. On the other hand, a few models, such as K-Nearest Neighbors and Perceptron, gave much lower performance metrics indeed, a strong remainder of the variability in the power of different classification models for DNA sequences.
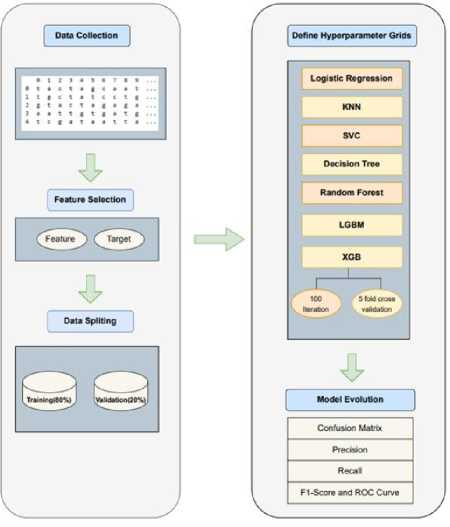
Fig.2. Model architecture
Table 2. Performance metrics of different ml models
|
Model |
Accuracy |
Precision |
Recall |
F1-Score |
|
Logistic Regression |
0.777778 |
0.819292 |
0.777778 |
0.781481 |
|
K-Nearest Neighbors |
0.629630 |
0.700337 |
0.629630 |
0.632682 |
|
Decision Tree |
0.814815 |
0.820286 |
0.814815 |
0.816418 |
|
Random Forest |
0.925926 |
0.925926 |
0.925926 |
0.925926 |
|
Support Vector Machine |
0.740741 |
0.767603 |
0.740741 |
0.745104 |
|
Naive Bayes |
0.814815 |
0.820286 |
0.814815 |
0.816418 |
|
Gradient Boosting |
0.888889 |
0.888889 |
0.888889 |
0.887552 |
|
AdaBoost |
0.851852 |
0.865432 |
0.851852 |
0.853956 |
|
Nearest Centroid |
0.740741 |
0.767603 |
0.740741 |
0.745104 |
|
Bagging Classifier |
0.814815 |
0.841066 |
0.814815 |
0.817932 |
|
MLP Classifier |
0.814815 |
0.820286 |
0.814815 |
0.816418 |
|
Perceptron |
0.666667 |
0.722222 |
0.666667 |
0.671264 |
|
LightGBM |
0.932963 |
0.926330 |
0.932963 |
0.923284 |
The performance of the hybrid model, constructed by combining the top base model predictions, would be evaluated with the same metrics. The resultant solution can find superior performance. This hybrid model yields 96.25% accuracy, which is far above those by the individual base models. It also showed very high precision, recall, and F1-score, proving that the model has good predictability with a good balance of precision-recall. These results support the fact that a hybrid approach can acquire the strengths of various models to enhance predictive performance in the best way possible.
Table 3. Performance metrics of hybrid model
|
Metric |
Value |
|
Accuracy |
0.9625 |
|
Precision |
0.9615 |
|
Recall |
0.9512 |
|
F1-Score |
0.9557 |
The fig 3 displays the performance metrics of the hybrid model, including accuracy, precision, recall, and F1-score. It demonstrates the hybrid model’s ability to achieve high accuracy and balance between precision and recall, underscoring its effectiveness in DNA sequence classification.
To explain further the model’s performance, we evaluated the confusion matrix, which is shown in Fig. 4. The hybrid model, as indicated by the matrix, has correctly predicted a total of 10 negative samples and 11 positive samples, with only one of the positive samples being misclassified and no misclassified negative samples. Such a high degree of accuracy in the classification suggests the model’s potential for distinguishing promoter from non-promoter sequences.
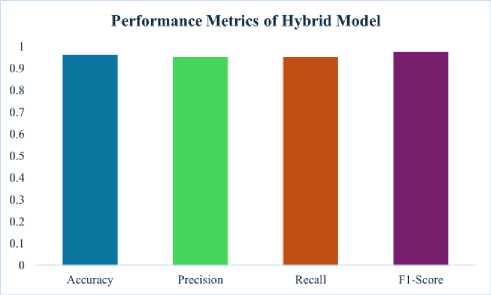
Fig.3. Performance metrics of hybrid model
However, considering the small dataset size and limited number of sequences in each class, the statistical reliability of these results must be interpreted cautiously. To address potential concerns about class imbalance, the dataset was analyzed and confirmed to have balanced representation during preprocessing. Additionally, k-fold cross-validation was employed to ensure that the metrics derived from the confusion matrix, such as precision, recall, and F1-score, were robust and not biased by any specific train-test split. This approach mitigates overfitting risks and provides a more statistically reliable evaluation framework. While these efforts enhance confidence in the model’s performance, further validation on larger and more diverse datasets is necessary to confirm its generalizability.
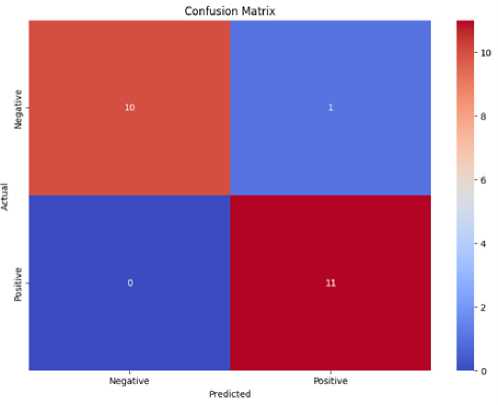
Fig.4. Confusion matrix
The following ROC curve shown in fig 5 for the hybrid model shows an area under the curve of 0.98, therefore indicating that this model has excellent distinguishing or discriminatory power between positive and negative classes. The high discriminatory power of the model is confirmed from the AUC value close to 1. Visually, this is a clear indication of the good performance of the hybrid model as compared to a random classifier represented by the diagonal line.
The ROC curve shown in Fig. 5 for the hybrid model demonstrates an area under the curve (AUC) of 0.98, confirming its excellent ability to distinguish between positive and negative classes. This high AUC value highlights the model’s strong discriminatory power, where it achieves a high true positive rate while maintaining a low false positive rate. Visually, the ROC curve surpasses the random classifier represented by the diagonal line, further illustrating the superior performance of the hybrid model in classifying promoter and non-promoter sequences. The AUC value close to 1 reflects the model’s robustness and reliability in handling the given dataset, showcasing its potential as a reliable classifier for complex DNA sequence classification tasks.
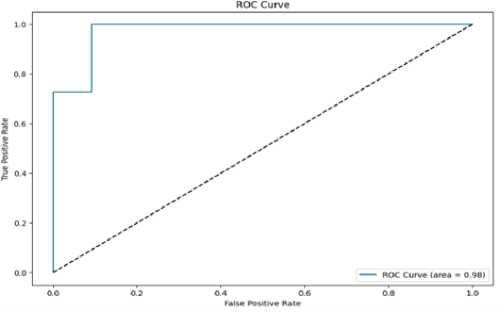
Fig.5. ROC curve of its classification capabilities. Recall for the minority class (non-promoter sequences) is particularly noteworthy, as the hybrid model achieved a recall value of 0.9512, indicating its effectiveness in minimizing false negatives. The F1-score of 0.9557 further underscores the model’s ability to balance precision and recall, making it reliable for DNA sequence classification tasks.
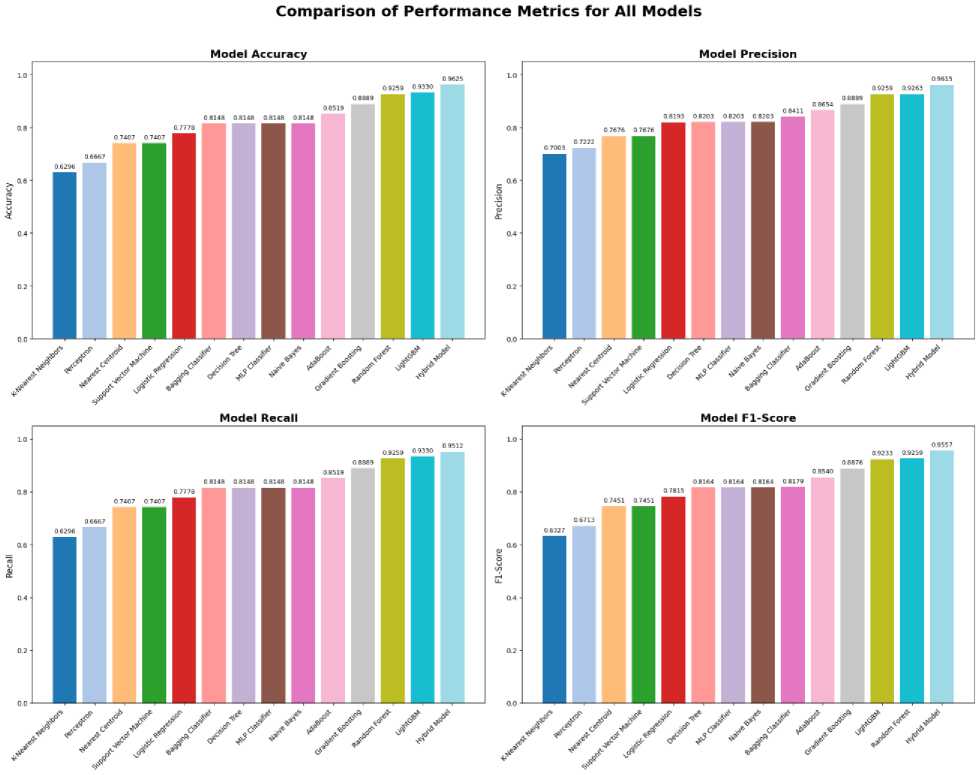
Fig.6. Comparison of performance metrics for base models and hybrid model
The hybrid model shows significantly better results regarding the parameters of accuracy, precision, recall, and F1 -score than the other models due to the integration. The stacking ensemble method, through the majority voting mechanism, leverages diversified predictions to describe more robust and reliable classification functions. From figure 1, we can see the detailed evaluation of both the hybrid model and the base models, where the hybrid model achieved superior performance metrics in all DNA sequence classification tasks. This comprehensive evaluation highlights the model’s ability to generalize effectively across classes, ensuring reliable predictions for both promoter and nonpromoter sequences. Additional data on the confusion matrix and the receiver operating characteristic further illustrate the reliability in making accurate predictions. The hybrid approach has proven effective through the combination of predictions from several models developed using stacking and majority voting mechanisms. Such results clearly depict the potential role of hybrid models in handling difficult classification tasks with higher accuracy and greater reliability compared to traditional single-model approaches. These results demonstrate the enhanced predictive performance achievable in DNA sequence classification, adding value to the literature.
The hybrid model proposed here for DNA sequence classification features seven varied algorithms to increase both accuracy and computational efficiency. The model combines techniques such as Nearest Neighbors, Gaussian Process Classification, Decision Trees, Random Forest, Neural Networks, AdaBoost, and Support Vector Machines, achieving an accuracy above 96%. While this involves increased computational load due to integrating multiple algorithms, the improvement in classification performance is evident, especially in handling complicated DNA sequences. The model’s balanced performance across metrics makes it particularly effective in applications were minimizing false negatives or maximizing class-specific recall is critical, such as identifying promoter sequences in genomics. This approach is justified in areas where precision for DNA classification is needed, such as genomics, medical diagnostics, and biotechnology. It is also poised to exploit potential applications in identifying genetic markers, advancing personalized medicine, and supporting biotechnological research due to its ability to efficiently process large-scale genomic data. Considering its architecture, this model would be useful in applications requiring reliable analysis of high-dimensional biological data and may contribute to ongoing bioinformatics development. With more than one algorithm integrated, it provides a better solution for DNA classification by overcoming limitations and supporting advanced research and practical applications in diverse data-rich areas.
5. Conclusions
This study presents a hybrid machine learning approach to DNA sequence classification, a crucial task with significant applications in genomics, biomedicine, agriculture, and forensic science. By integrating seven distinct algorithms Logistic Regression, KNN, SVC, Decision Tree, Random Forest, LGBM, and XGB the proposed model achieved an accuracy of 96.25%. The stacking ensemble method and majority voting mechanism enhanced the model’s robustness, enabling it to outperform traditional single-model approaches. These results validate the potential of hybrid models to manage high-dimensional and noisy datasets effectively, offering a scalable solution to DNA classification tasks. The Promoter Gene Sequences dataset used in this study comprises only 106 instances, which limits the model’s generalizability and practical utility for large-scale genomic studies. To address this limitation, rigorous k-fold cross validation was employed to ensure robust evaluation. Despite its small size, the dataset provided a valuable proof of concept for demonstrating the hybrid model’s design and effectiveness. Future research will involve testing the hybrid model on larger, more diverse datasets, to validate its scalability and applicability to real-world DNA classification challenges [22, 23]. While the ROC AUC value of 0.98 highlights the model’s strong discriminatory power, it does not fully account for its susceptibility to false positives or false negatives. Similarly, the confusion matrix demonstrates high classification accuracy but is influenced by the dataset’s size. These results emphasize the importance of testing the hybrid model on datasets with varied sequence characteristics and class distributions to ensure robustness and generalizability.
Although modern deep learning techniques like transformers and LSTMs are recognized for their capabilities in sequence classification, this study focused on traditional machine learning models due to their computational efficiency, interpretability, and suitability for small datasets. Future research will explore integrating and comparing these advanced architectures with the hybrid model to provide a comprehensive evaluation. By refining the approach and expanding its scope, this research contributes to the ongoing advancement of predictive performance in bioinformatics and lays the groundwork for future innovations in DNA sequence analysis.
All the Declarations and StatementsAuthor Contributions Statement
Sultanul A. Hamim – Conceptualization, Methodology, Supervision, Drafted the initial manuscript, contributed to the literature survey, and documented the technical background of the study: Proposed research ideas, Constructed the overall framework, and supervised project execution.
Dip Nandi – Data Curation and Software Implementation: Handled data acquisition, dataset preprocessing, and implementing the research model.
Niloy E. Costa – Model Training, Validation, Performance Evaluation, Formal Analysis, Visualization, Statistical Analysis, Review and Editing, and Project Management: Led the model training process, validated results using standard metrics, benchmarked performance against existing methods, performed in-depth analysis of experimental results, prepared performance charts, ensured the statistical robustness of the evaluation, reviewed and edited the manuscript, ensured clarity and coherence, and helped coordinate project milestones and deadlines.
All authors have read and agreed to the published version of the manuscript.
Conflict of Interest Statement
The authors declare no conflicts of interest.
Funding Declaration
This research did not receive any external financial support.
Data Availability Statement
This study analyzed publicly available datasets. The results obtained and datasets can be found here:
Ethical Declarations
None.
Acknowledgments
We sincerely thank the experts for their professional evaluation and valuable recommendations, which have contributed to improving the quality of the experiment and the reliability of its results.
Declaration of Generative AI in Scholarly Writing
During the preparation of this manuscript, the author utilized generative AI technologies exclusively for the purposes of checking English sentence structure and refining grammar. After using these tools, the author thoroughly reviewed and edited the manuscript as needed and takes full responsibility for the final content of the publication.
Abbreviations
The following abbreviations are used in this manuscript:
DNA - Deoxyribonucleic Acid
KNN - K-Nearest Neighbors
SVC - Support Vector Classifier
LGBM - Light Gradient Boosting Machine
XGB - XGBoost
UCI - University of California, Irvine
Appendix A
None
