Идентификация клинически значимых вариантов генов эмбрионального развития сердечно-сосудистой системы при спорадической аневризме восходящего отдела и «бычьей дуге» аорты

Автор: Гончарова И.А., Панфилов Д.С., Слепцов А.А., Шипулина С.А., Зарубин А.А., Бабушкина Н.П., Валиахметов Н.Р., Саушкин В.В., Лелик Е.В., Петракова Е.А., Козлов Б.Н., Назаренко М.С.
Журнал: Сибирский журнал клинической и экспериментальной медицины @cardiotomsk
Рубрика: Клинические исследования
Статья в выпуске: 4 т.40, 2025 года.
Бесплатный доступ
Введение. Аномалии дуги аорты, в большей степени «бычья дуга» аорты, могут обусловливать развитие аневризмы восходящей аорты. Наблюдается высокий коэффициент наследуемости данной патологии, однако молекулярно-генетические исследования единичны. Поскольку «бычья дуга» является одним из вариантов развития дуги аорты и магистральных сосудов в процессе эмбриогенеза, возможно, что данная аномалия связана с генами, кодирующими белки, вовлеченные в эмбриональное развитие сердечно-сосудистой системы. Цель: поиск редких клинически значимых вариантов генов эмбрионального развития сердечно-сосудистой системы у больных со спорадической аневризмой восходящей аорты с дугой аорты нормального строения и в сочетании с «бычьей дугой». Материал и методы. В исследование включены 42 пациента со спорадической формой аневризмы восходящей аорты, в том числе 11 человек с «бычьей дугой» аорты. Анализ клинического экзома выполнялся на основании данных секвенирования ДНК-библиотек Clinical Exome Solution (Sophia Genetics, Швейцария) на приборе NextSeq 500 (Illumina, США). Поиск редких клинически значимых вариантов (частота минорного аллеля < 1%) проводился в экзонах и прилегающих к ним интронах 120 генов эмбрионального развития сердечно-сосудистой системы. Валидация выявленных вариантов выполнялась секвенированием по Сэнгеру. Результаты. У больных с аневризмой аорты и «бычьей дугой» выявлены следующие клинически значимые варианты: патогенный вариант c.610-2A-G гена CCDC39, который представляет собой однонуклеотидную замену, приводящую к потере акцепторного сайта сплайсинга (ΔScore = 0,97 Spliceailookup), и вариант с неопределенным клиническим значением (VUS – variant of uncertain clinical significance) c.2564T-C в гене ANKS6, имеющий высокие показатели патогенности по шкалам CADD (Phred = 28,3) и AlphaMissense (0,972). В группе больных с аневризмой аорты с нормальной анатомией супрааортальных сосудов выявлен вероятно патогенный вариант с.1151T-C гена ACVR2B (AlphaMissense = 0,966). Среди 38 генов, в последовательности которых обнаружены VUS в обеих группах больных, белковые продукты 17 (44,7%) вовлечены в функционирование ресничек и микротрубочек, а белки, кодируемые генами MKS1, CCDC40, DNAAF1, ANKS6, CCDC39, DNAH5, DNAAF3, отвечают и за развитие сердечно-сосудистой системы. Заключение. В развитие спорадической формы аневризмы восходящей аорты в сочетании с «бычьей дугой» вносят вклад редкие клинически значимые варианты генов CCDC39 и ANKS6, ответственные за функционирование первичных ресничек. При наличии дуги нормального строения важную роль играют варианты гена ACVR2B, принадлежащего к суперсемейству сигнальных белков TGF-бета.
Аневризма восходящей аорты, «бычья дуга», клинически значимые генетические варианты, гены эмбрионального развития сердечно-сосудистой системы, CCDC39, ANKS6, ACVR2B
Короткий адрес: https://sciup.org/149150143
IDR: 149150143 | УДК: 616.132.14-007.64:616.1:577.212 | DOI: 10.29001/2073-8552-2025-40-4-81-89
Identification of clinically significant variants in genes of cardiovascular embryonic development in sporadic ascending aortic aneurysm and “bovine aortic arch”
Introduction. Aortic arch anomalies, especially the “bovine arch”, can cause the development of an ascending aortic aneurysm. There is a high coefficient of heritability of this pathology, however, genetic studies are rare. Since the “bovine arch” is one of the variants of the development of the aortic arch and large vessels during embryogenesis, this pathology may be associated with genes encoding proteins involved in the embryonic development of the cardiovascular system. Aim: To identify rare, clinically significant variants of genes of cardiovascular embryonic development in patients with sporadic ascending aortic aneurysm and a “bovine arch”. Material and Methods. The study included 42 patients with a sporadic form of ascending aortic aneurysm, including 11 patients with a “bovine arch”. Analysis of the clinical exome was performed based on DNA sequencing data using Clinical Exome Solution (Sophia Genetics, Switzerland) and NextSeq 500 genetic sequencer (Illumina, USA). The search for rare, clinically significant variants (minor allele frequency <1%) was carried out in exons and adjacent introns of 120 genes of embryonic development of the cardiovascular system. Validation of identified variants was performed using Sanger sequencing. Results. In patients with aortic aneurysm and “bovine arch”, the following clinically significant variants were identified: the pathogenic variant c.610-2A-G of the CCDC39 gene, which is a single-nucleotide substitution leading to the loss of the acceptor splice site (ΔScore = 0.97 Spliceailookup) and a variant of uncertain clinical significance (VUS) c.2564T-C in the ANKS6 gene, which has high pathogenicity rates on the CADD (Phred = 28.3) and AlphaMissense (0.972) scales. A likely pathogenic variant c.1151T-C of the ACVR2B gene was identified in the group of patients with aortic aneurysm without supraaortic vessels anomaly (AlphaMissense = 0.966). Among the 38 genes whose sequences revealed VUS in both groups of patients, the protein products of 17 (44.7%) are involved in the functioning of cilia and microtubules, and the proteins encoded by the genes MKS1, CCDC40, DNAAF1, ANKS6, CCDC39, DNAH5, DNAAF3 are also responsible for the development of the cardiovascular system. Conclusion. Rare, clinically significant variants in the CCDC39 and ANKS6 genes, which are crucial for primary cilia function, contribute to the development of sporadic ascending aortic aneurysm in combination with a “bull’s arch.” When a normal aortic arch is present, variants in the ACVR2B gene, belonging to the TGF-beta signaling protein superfamily, play an important role.
Текст научной статьи Идентификация клинически значимых вариантов генов эмбрионального развития сердечно-сосудистой системы при спорадической аневризме восходящего отдела и «бычьей дуге» аорты
Предрасположенность к развитию спорадической аневризмы восходящей аорты формируется по принципу, отражающему формирование большинства многофакторных заболеваний и сложно наследуемых признаков, где вклад в развитие патологии вносят как генетические причины, так и целый спектр различных факторов риска.
По некоторым данным, аномалии дуги аорты, в большей степени формирование общего ствола брахиоцефальной и плечеголовной артерий или «бычья дуга» (CILCA – common origin of the innominate and left carotid artery), могут обусловливать развитие аневризмы восходящей аорты [1]. И если популяционная частота CILCA колеблется в пределах 10–26%, то у пациентов с заболеваниями восходящей аорты она достигает 35% [2]. Показано семейное накопление данной патологии до 53% и высокий коэффициент наследуемости (h2 = 0,71) [3]. Вместе с тем к настоящему времени попытки выявить генетические причины развития «бычьей дуги» практически не предпринимались. В одной из работ идентифицированы варианты неопределенной клинической значимости (VUS) в генах FBN2, COL5A2 и NOTCH1 в семьях с аневризмой восходящей аорты и «бычьей дугой» [3]. Поскольку данные варианты локализованы в генах наследственных аневризм аорты, они в большей степени связаны с развитием аневризмы, а не CILCA.
«Бычья дуга» является одним из вариантов развития дуги аорты и магистральных сосудов в процессе эмбриогенеза. Возможно, что генетическая основа предрасположенности к данной патологии затрагивает гены, кодирующие белки, вовлеченные в эмбриональное развитие сердечно-сосудистой системы, ангиогенез, ремоделирование внеклеточного матрикса, формирование цитоскелета и цилиарного аппарата.
В частности, экспериментальные исследования на модельных животных показали, что нарушение эмбрионального развития и формирование врожденных пороков сердца и сосудов связано и с функционированием первичных ресничек, которые играют ключевую роль в поддержании гомеостаза сердечно-сосудистой системы [4]. Первичные реснички функционируют как клеточные «антенны» для координации сигнальных путей Notch1, Hedgehog, Wnt, TGF-beta, как механосенсоры на эндотелиальных клетках и регуляторы ремоделирования внеклеточного матрикса [5, 6]. Как было показано ранее, нарушение функционирования первичных ресничек связано с нарушением процесса латерализации, развитием врожденных пороков сердца, аномалиями митрального и аортального клапанов, а также изменением механочувствительности тканей и развитием патологических изменений при гемодинамической нагрузке [7]. При аневризме восходящей аорты, как известно, активируются сигнальные пути Notch1, Hedgehog, Wnt, TGF-beta, происходит ремоделирование внеклеточного матрикса [8, 9]. Таким образом, функционирование цилиарного аппарата связано не только с развитием врожденных пороков сердца и клапанов, но и, возможно, с формированием «бычьей дуги» и впоследствии аневризмы аорты.
Цель исследования: поиск редких клинически значимых вариантов генов эмбрионального развития сердечно-сосудистой системы у больных со спорадической аневризмой восходящей аорты с дугой аорты нормального строения и в сочетании с «бычьей дугой».
Материал и методы
Исследование проведено в соответствии с Хельсинкской декларацией и Надлежащей клинической практикой (GCP), одобрено локальным комитетом по биомедицинской этике НИИ медицинской генетики Томского НИМЦ (протокол № 13 от 15 ноября 2021 г.) и локальным комитетом по биомедицинской этике НИИ кардиологии Томского НИМЦ (протокол № 213 от 12 мая 2021 г.).
В исследование включены 42 пациента со спорадической формой аневризмы восходящей аорты, среди которых 11 характеризовались наличием «бычьей дуги» аорты, остальные пациенты имели дугу аорты нормального строения. Всем пациентам проведено открытое хирургическое лечение в НИИ кардиологии Томского НИМЦ в 2020–2023 гг. Критериями включения пациентов в исследование были: аневризма аорты с расширением восходящей части ≥ 5,0 см без сочетанного поражения аортального клапана; дилатация аорты с расширением восходящей части ≥ 4,5 см при выраженном наличии стеноза аортального клапана или при его недостаточности выше второй степени. Из исследования исключены пациенты с расслоением / разрывом аорты, с ложными аневризмами аорты, а также те, которым требуются повторные вмешательства на проксимальных отделах грудной аорты.
Размеры аорты определяли с помощью мультиспи-ральной компьютерной томографии (МСКТ) на томографе GE Discovery NM/CT 570С (GE Healthcare, США) со следующими параметрами: 200–400 мА, 100–120 кВт. Полученные изображения обрабатывали на специализированной рабочей станции Advantage Workstation 4.3 (GE Healthcare, США).
Взятие венозной крови (5–10 мл) проводил сертифицированный медицинский персонал в стерильные одноразовые вакуумные пробирки с ЭДТА-К2 Improvacuter (Guangzhou Improve Medical Instruments Co., Ltd, Китай). Образцы крови замораживали и хранили при температуре –20 ºС.
Из лейкоцитов периферической крови с помощью фенол-хлороформной экстракции проводили выделение ДНК, которую растворяли в TE-буфере. Качество выделенной ДНК оценивали на спектрофотометре Nanodrop 8000 (Thermo, США) и в 1% агарозном геле c 1X ТАЕ-бу-фером. Количественную оценку ДНК осуществляли с помощью флуориметра Qubit 3 (Thermo, США).
ДНК-библиотеки создавали при помощи набора Clinical Exome Solution (Sophia Genetics, Швейцария), KAPA Library Amplification kit KK2620 (Roche, Германия) и магнитных частиц Agencourt AMPure XP (Beckman Coulter, США) по протоколу Clinical Exome Solution (Sophia Genetics, Швейцария). Анализ качества ДНК-библиотек выполняли на флуориметре Qubit 3 (Thermo, США) и системе капиллярного электрофореза Bioanalyzer 2100 (Agilent, США), согласно протоколам производителей. Секвенирование ДНК-библиотек проводили на приборе NextSeq 500 (Illumina, США) с набором NextSeq 500/550 Mid Output Kit v2, согласно протоколу производителя. Прочтения коротких фрагментов ДНК выравнивали на референсный геном (GRCh38) с помощью Illumina DRAGEN Bio-IT (Illumina, США). Аннотацию генетических вариантов выполняли в программе ANNOVAR.
Классификацию идентифицированных вариантов по степени патогенности проводили согласно стандартам и рекомендациям Американского колледжа медицинской генетики и геномики (ACMG, the American College of Medical Genetics and Genomics) и Ассоциации молекулярной патологии (the Association for Molecular Pathology) по интерпретации вариантов последовательностей (Standards and guidelines for the interpretation of sequence variants), с использованием инструментов VarSome The Human Genomics Community (включая in silico предикторы (MutPred, MVP, PrimateAI, EIGEN PC, LIST-S2, LRT, PROVEAN, SIFT)1 , InterVar2 и Franclin3. Оценку влияния миссенс вариантов на патологическое изменение структуры белка проводили с помощью инструмента AlphaMissense4. Оценку влияния вариантов на изменение сплайсинга выполняли с помощью инструмента Spliceailookup5 на основании показателя ΔScore (показатель изменения сплайсинга на основании оценки разницы между границами экзон-интрон для референсной и альтернативной последовательности транскриптов). Связывание транскрипционных факторов (ТФ) в сайтах локализации клинически значимых генетических вариантов оценивали согласно предикции базы данных JASPAR, результаты которой представлены в геномном браузере
UCSC6. Частоту генетических вариантов в популяциях определяли с помощью информации в базах данных RUSeq7 и gnomAD v. 4.1.0.8 .
Поиск редких генетических вариантов (частота минорного аллеля < 1%) проводили в экзонах и прилегающих к ним интронах 120 генов эмбрионального развития сердечно-сосудистой системы, куда входят гены, вовлеченные в ангиогенез, ремоделирование внеклеточного матрикса, формирование цитоскелета и цилиарного аппарата. Выбор генов сделан на основании данных, представленных в научной литературе [7, 10].
Валидацию клинически значимых вариантов генов CCDC39 и ACVR2B выполняли двунаправленным секвенированием по Сэнгеру на генетическом анализаторе 3730xl DNA Analyzer (Applied Biosystems, USA). ПЦР-ам-плификацию проводили с использованием готовой реакционной смеси HS-Taq PCR-Color (2×) (Биолабмикс, Россия) с праймерами, указанными в таблице 1.
Таблица 1 . Гены и праймеры, использованные для секвенирования по Сэнгеру
Table 1 . Genes and primers used for Sanger sequencing
|
Ген |
Локализация варианта |
Последовательность праймеров |
|
CCDC39 |
Интрон 5 |
F: 5’- TGGATATAGAGTCAGCTTTCA -3’ R: 5’- GCACTGACTCTGCAATTAG -3’ |
|
ACVR2B |
Экзон 9 |
F: 5’- CTGTCTGAATTGCTCTGTC -3’ R: 5’- GCCAATCTCTTCCTCAAAG -3’ |
Праймеры подобраны с помощью инструментов Primer Quest Tool9 в соответствии с последовательностью референсного генома GRCh38. Для очистки продуктов ПЦР использовали термолабильную экзонуклеазу Exo I (Thermo Scientific, США) и щелочную фосфатазу креветок (Thermo Scientific, США). Последующую реакцию секвенирования ДНК проводили с использованием набора BigDye Sequencing Kit v3.1 (Thermo Scientific, США), 5-кратного буфера для секвенирования, прямого или обратного праймера, продукта ПЦР. Продукты реакции секвенирования очищали с помощью набора D-Pure Reagent Kit (Nijmegen, Нидерланды). Визуализацию и анализ результатов секвенирования ДНК проводили в программном обеспечении UGENE (v42.0, Unipro, Рос-сия)10.
Количественные показатели пациентов представлены средним значением ( М ) и стандартным отклонением ( SD ), М ± SD . Категориальные показатели описаны частотами встречаемости, n (%). Оценку значимости различий категориальных признаков в двух независимых группах проводили с помощью χ 2 -критерия Пирсона, при оценке количественных показателей использовали критерий Стьюдента. Критический уровень значимости при проверке гипотезы о различиях показателей в двух независимых группах составлял 0,05.
Результаты
Среди 42 пациентов с аневризмой восходящего отдела, включенных в исследование, было 28 мужчин (средний возраст – 55,7 ± 11,6 года) и 14 женщин (средний возраст – 57,7 ± 11,6 года). Одиннадцать пациентов характеризовались наличием CILCA. Группы пациентов без аномалий дуги аорты и с «бычьей дугой» были сопоставимы по основным клиническим показателям (табл. 2).
В 120 генах эмбрионального развития сердечно-сосудистой системы у 42 пациентов с аневризмой восходящего отдела выявлено 6346 генетических вариантов, включающих короткие инсерции / делеции, синонимичные / несинонимичные однонуклеотидные замены и варианты, приводящие к образованию стоп-кодона или нарушающие сайт сплайсинга. В анализ вошли миссенс-замены, влияющие на аминокислотную последовательность белка, варианты, приводящие к образованию стоп-кодона или нарушающие сайт сплайсинга, распространенные в популяциях с частотой менее 1% (по данным gnomAD v. 4.1.0 и RUSeq), или частота минорного аллеля которых неизвестна. Дополнительным критерием выбора вариантов явилась глубина прочтения, которая составила не менее 30 прочтений на образец.
После установления вышеописанных критериев остался 121 вариант. Данные варианты были классифицированы по степени патогенности, в результате чего идентифицированы следующие варианты: патогенный в гене CCDC39 (c.610-2A>G); неоднозначно классифицированный в гене ACVR2B (c.1151T>C:p.Ile384Thr) (вероятно доброкачественный – VarSome; вариант неясного клинического значения (VUS) – Franklin); вероятно патогенный –InterVar; табл. 3); 67 VUS; 52 вероятно доброкачественных или доброкачественных.
Патогенный вариант гена CCDC39 – c.610-2A>G, выявленный в гетерозиготном состоянии у пациента с аневризмой восходящей аорты и CILCA, представляет собой однонуклеотидную замену, приводящую к потере акцепторного сайта сплайсинга (ΔScore = 0,97; см. табл. 3, рис. 1). Он локализован в сайте связывания ТФ MEIS3 и PKNOX1.
Вариант c.1151T>C гена ACVR2B , выявленный в гетерозиготном состоянии у пациента с дилатацией без аномалии строения дуги аорты, является миссенс-вари-антом, приводящим к замене аминокислот p.Ile384Thr (см. табл. 3, рис. 1). Согласно классификации ACMG-AMP, он имеет неоднозначный статус (LB, VUS, LP). Од-
Таблица 2 . Клиническое описание пациентов, включенных в исследование
Table 2 . Clinical description of patients included in the study
|
Характеристики |
Вся группа, n = 42 |
Без аномалий дуги, n = 31 |
С «бычьей дугой», n = 11 |
p |
|
Возраст, лет |
55,6 ± 11,5 |
57,4 ± 11,1 |
52,1 ± 12,4 |
0,22 |
|
Диаметр восходящей аорты |
52,2 ± 7,3 |
52,9 ± 8,3 |
50,4 ± 3,2 |
0,52 |
|
Диаметр корня аорты |
44,1 ± 9,0 |
43,6 ± 9,0 |
45,6 ± 9,4 |
0,32 |
|
Диаметр проксимальной части дуги аорты |
39,2 ± 4,4 |
39,5 ± 3,8 |
38,5 ± 5,9 |
0,77 |
|
Двустворчатый аортальный клапан, n (%) |
25 (59,5) |
20 (64,5) |
5 (45,4) |
0,27 |
|
Артериальная гипертензия, n (%) |
27 (64,3) |
20 (64,5) |
7 (63,6) |
0,96 |
|
Гиперлипидемия, n (%) |
10 (23,8) |
8 (25,8) |
2 (18,1) |
0,61 |
|
Ожирение (ИМТ > 30), n (%) |
18 (42,8) |
12 (38,7) |
6 (54,5) |
0,36 |
|
СД2 или нарушение толерантности к глюкозе, n (%) |
3 (7,1) |
2 (6,4) |
1 (9,1) |
0,77 |
|
Стенокардия, ИБС или инфаркт миокарда, n (%) |
14 (33,3) |
11 (35,5) |
3 (27,3) |
0,62 |
Примечание: ИМТ – индекс массы тела; СД2 – сахарный диабет второго типа; ИБС – ишемическая болезнь сердца.
нако большая часть индивидуальных in silico предикторов (MutPred, MVP, PrimateAI, EIGEN PC, LIST-S2, LRT, PROVEAN, SIFT) свидетельствует в пользу его умерен- ной патогенности. Инструмент AlphaMissense, предсказывающий влияние миссенс-мутаций на синтез структурно измененного, связанного с патологическими состояниями
Таблица 3 . Клинически значимые генетические варианты, выявленные у пациентов с аневризмой восходящей аорты
Table 3 . Clinically significant genetic variants identified in patients with ascending aortic aneurysm
|
Клинические и генетические характеристики |
Пациент № 14 |
Пациент № 17 |
|
Клинический диагноз |
Супракоронарная аневризма, «бычья дуга» |
Дилатация аорты |
|
Пол |
ж |
м |
|
Возраст |
51 |
68 |
|
Диаметр аорты, мм, по MСКТ1 |
44,6/52,7/36 |
40/42/44 |
|
АоК |
БАК |
ТрАК |
|
Факторы риска сердечно-сосудистых заболеваний (АГ / СД2 / ГПЛ / ожирение) |
нет/нет/нд/нет |
да/нет/нд/нет |
|
Ген, координаты (GRCh38) (Ref/Alt), замена нуклеотидов: аминокислот |
CCDC39, chr3:180659582 (T/C), c.610-2A>G: нет |
ACVR2B, chr3:38482274 (Т/С), c.1151T>C: p.Ile384Thr |
|
Идентификационный номер варианта (частота минорного аллеля2); Патогенность3 |
rs756235547 (1,4х10-4 /1,1х10-4); Р/P/нд, 32 |
rs1249314121 (1,9х10-5/5х10-4); LB/VUS/LP, 28,7 |
Примечание: м – мужчина; ж – женщина; АоК – строение аортального клапана; БАК – двустворчатый аортальный клапан; ТрАК – трехстворчатый аортальный клапан; нд – нет данных; АГ – артериальная гипертензия; СД2 – сахарный диабет второго типа; ГПЛ – гиперлипидемия; Р – патогенный генетический вариант; Ref – референсный аллель; Alt – альтернативный аллель; LP – вероятно патогенный генетический вариант; VUS – вариант с неопределенным клиническим значением; LB – вероятно доброкачественный генетический вариант; 1 диаметры аорты в областях синотубулярного соединения / средней части восходящей аорты / проксимальной дуги аорты по результатам мультиспиральной компьютерной томографии (МСКТ); 2 – частота минорного аллеля по gnomAD (Eur)/ Ruseq; 3 – оценка патогенности по шкалам предикции патогенности Varsome / Franklin / Intervar, CADD (Phred).
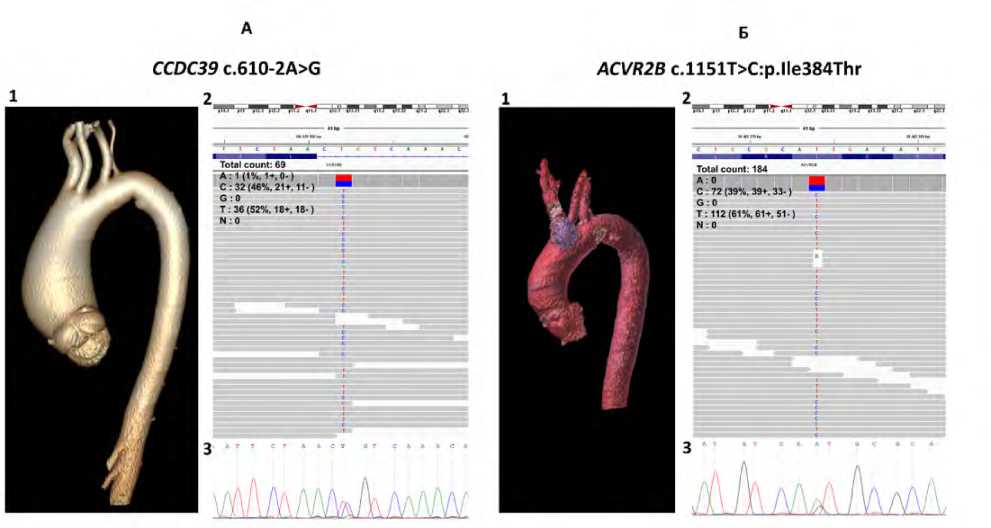
Рис. 1. Выявленные генетические варианты у пациентов с аневризмой восходящего отдела. (A): 1 – МСКТ грудной аорты у пациента № 14, 2 – результаты молекулярно-генетического тестирования методом секвенирования клинического экзома, 3 – валидация секвенирования по Сэнгеру варианта chr3:180659582 (T/C), c.610-2A>G в гене CCDC39. (Б): – МСКТ грудной аорты у пациента № 17, 2 – результаты молекулярно-генетического тестирования методом секвенирования клинического экзома, 3 – валидация секвенирования по Сэнгеру варианта chr3:38482274 (Т/С), c.1151T>C:p. Ile384Thr гена ACVR2B
Fig. 1. Identified genetic variants in patients with ascending aorta aneurysm. (A): 1 – thoracic aorta MSCT in patient №14, 2 – results of molecular genetic testing by clinical exome sequencing, 3 –Sanger sequencing validation of the chr3:180659582 (T/C) c.610-2A>G variant in the CCDC39 gene. (B): – thoracic aorta MSCT in patient №17, 2 – results of molecular genetic testing by clinical exome sequencing, 3 – Sanger sequencing validation of the chr3:38482274 (T/C) c.1151T>C:p.Ile384Thr variant in the ACVR2B gene
белка, также относит данный вариант к классу вероятно патогенных (балл патогенности 0,966).
Как было отмечено выше, у больных с аневризмой восходящей аорты выявлено 67 VUS в 38 генах, из них 57 вариантов (36 генов) при нормальном строении дуги аорты и 13 вариантов (12 генов) при наличии «бычьей дуги». Часть вариантов или генов перекрывается между группами. Так, три VUS генов HYDIN (c.12076G>A), HOXA3 (c.1127G>A) и COQ9 (c.826C>T) являются общими для обеих групп больных. У пациентов с дугой аорты различного строения выявлены разные генетические варианты одних и тех же генов MEGF8, MKS1, GALNT11, DNAH9, DNAH5, KMT2D, CCDC40 . А VUS в генах ANKS6 (c.2564T>C:p.Phe855Ser (rs200644058)) и FBN1 (c.7841C>T:p.Ala2614Val) обнаружены только у больных с «бычьей дугой».
Несмотря на то, что c.2564T>C гена ANKS6 классифицирована как VUS (InterVar) или вероятно доброкачественный (Varsom/Franklin) вариант, данная замена имеет показатель CADD (Phred = 28,3), что относит ее к числу высокопатогенных вариантов. Инструмент AlphaMissense присваивает данному варианту балл патогенности 0,972 и относит его к вероятно патогенным. Данный вариант локализован в последовательности цис-регуляторного элемента EH38E2711785 и сайте связывания ТФ ElF5. VUS в гене фибриллина FBN1 (c.7841C>T:p.Ala2614Val) ранее описан в данной группе больных при анализе клинически значимых генетических вариантов в генах наследственных аневризм восходящей аорты [11].
Среди 38 генов, в последовательности которых обнаружены VUS в обеих группах больных, белковые продукты 17 (44,7%) вовлечены в функционирование ресничек и микротрубочек, а белки, кодируемые генами MKS1,
CCDC40, DNAAF1, ANKS6, CCDC39, DNAH5, DNAAF3 , отвечают в том числе и за развитие сердечно-сосудистой системы. При сочетании аневризмы восходящей аорты и «бычьей дуги» количество генов с клинически значимыми генетическими вариантами, связанных с функционированием ресничек и микротрубочек, достигает 50% ( ANKS6, DNAH5, MKS1, HYDIN, DNAH9, CCDC40, CCDC39 из 12 генов, в которых выявлены генетические варианты).
Обсуждение
В настоящем исследовании был проведен поиск клинически значимых генетических вариантов в генах эмбрионального развития сердечно-сосудистой системы, белковые продукты которых вовлечены в ангиогенез, ремоделирование внеклеточного матрикса, формирование цитоскелета и цилиарного аппарата у больных с аневризмой восходящей аорты с наличием / отсутствием «бычьей дуги».
Белок, кодируемый геном CCDC39 , необходим для сборки динеинового регуляторного комплекса и важен для обеспечения подвижности ресничек эпителия. Ген CCDC39 входит в панель для диагностики цилиарной дискинезии – наследственного заболевания, характеризующегося рецидивирующими инфекциями верхних и нижних дыхательных путей, снижением фертильности у мужчин и зачастую зеркальным расположением внутренних органов (гетеротаксия) [12]. Патогенные варианты в гомозиготном или компаунд-гетерозиготном состоянии в этом гене связаны с развитием цилиарной дискинезии, гетеротаксии и врожденных пороков сердца [13].
В данном исследовании в гетерозиготном состоянии выявлен патогенный вариант c.610-2A>G гена CCDC39 , приводящий к потере акцепторного сайта сплайсинга
(ΔScore = 0,97). Размер эффекта генетических вариантов сплайсинга тесно связан с их баллами ΔScore, которые варьируют от 0 до 1. И чем выше балл прогнозирования модели, тем выше патогенный потенциал варианта изменения сплайсинга. Выявленный вариант также является сайтом связывания ТФ MEIS3 и PKNOX1, принимающих участие в развитии и функционировании сердечно-сосудистой системы [14, 15].
Вариант c.610-2A>G гена CCDC39 ранее был обнаружен у пациентов с цилиарной дискинезией и дефектами жгутика сперматозоидов [16]. У больного с аневризмой восходящей аорты и «бычьей дугой» этот вариант выявлен впервые. В анамнезе пациента нет гетеротаксии и врожденных пороков сердца, не сообщается и о других патологиях, развивающихся при наличии клинически значимых вариантов в гене CCDC39 . Это может быть обусловлено несколькими причинами. С одной стороны, вариант c.610-2A>G гена CCDC39 может быть связан с развитием «бычьей дуги» аорты, чего не было показано ранее. С другой стороны, выявленный патогенный вариант у данного пациента может характеризоваться неполной пенетрантностью – состоянием, когда одна и та же мутация вызывает развитие различных фенотипов, от отсутствия клинического проявления до тяжелого заболевания у носителей. Кроме этого, вариант c.610-2A>G гена CCDC39 может не быть «причинным» для развития «бычьей дуги», а может являться модификатором для другого генетического варианта, может изменять его пенетрантность и влиять на риск развития, манифестацию или тяжесть развития заболевания. В связи с этим на данном этапе можно лишь предполагать, что вариант c.610-2A>G гена CCDC39 связан с развитием «бычьей дуги». Для подтверждения этого факта необходимо проведение экспериментальных функциональных исследований, а также поиск клинически значимых вариантов в других группах генов.
У больных с аневризмой аорты и «бычьей дугой» выявлено 13 VUS. Несмотря на то, что VUS не включаются в заключение о результатах генетического тестирования, с появлением результатов исследований функциональной значимости данные варианты могут переходить из одной категории патогенности в другую, становиться вероятно патогенными или патогенными, что требует периодической переоценки. Из выявленных у пациентов с «бычьей дугой» VUS набольший интерес представляют миссенс-замены c.2564T>C:p.Phe855Ser гена ANKS6 и c.7841C>T:p.Ala2614Val гена FBN1 .
Ген ANKS6 кодирует белок, образующий первичные реснички. Клинически значимые варианты данного гена в гомозиготном или компаунд-гетерозиготном состоянии могут приводить к полиорганным порокам развития, нарушениям лево-правой асимметрии, поликистозу почек и врожденным порокам сердца, то есть патологиям, объединяемым в диагноз цилиопатий, связанных с неф-ронофтизом [17]. Выявленный в настоящем исследовании VUS c.2564T>C:p.Phe855Ser гена ANKS6 ранее был описан при поликистозе почек в сочетании с вариантами генов PKD2 и PKHD1 [18]. Ген PKD2 включен в панель генов, проанализированных в данной работе, однако в его последовательности у больных с аневризмой восходящей аорты не было выявлено клинически значимых вариантов. Вместе с тем поликистоз почек и нефроно-фтиз, являющиеся причиной хронической почечной недостаточности, могут сочетаться с недостаточностью клапанов сердца, митральным и аортальным стенозом и аневризмой аорты [19]. У пациента с выявленным в данном исследовании вариантом c.2564T>C:p.Phe855Ser гена ANKS6 наблюдается сочетание аневризмы восходящей аорты, патологии дуги аорты, стеноза аортального клапана и хронической болезни почек. Данный генетический вариант имеет высокие показатели патогенности по классификаторам CADD (Phred = 28,3) и AlphaMissense (0,972), что говорит о его высоко вероятном вкладе в развитие патологии. Кроме этого, данный вариант локализован в последовательности цис-регуляторного элемента EH38E2711785, влияющего на транскрипционную активность генов, расположенных на одной с ним хромосоме, и сайте связывания ТФ ElF5, важность которого для функционирования клеток сердечно-сосудистой системы и почек была показана в экспериментальных исследованиях [20].
При сочетании аневризмы восходящей аорты и «бычьей дуги» среди выявленных клинически значимых генетических вариантов 50% относятся к генам, белковые продукты которых влияют на функционирование первичных ресничек и микротрубочек. Экспериментальные исследования показали, что нормальное функционирование первичных ресничек играет решающую роль в развитии сосудов и поддержании гомеостаза сердечно-сосудистой системы. Нарушение подвижности или потеря эндотелиальных ресничек в эмбриогенезе ведет к нарушению сигнализации TGF-β, артериовенозной мальформации и формированию врожденных пороков сердца и сосудов [4].
Вариант c.7841C>T гена FBN1 имеет меньшие показатели патогенности CADD (Phred = 23,6) и AlphaMissense (0,112); он является миссенс-вариантом (p.Ala2614Val). У пациентов с аневризмой восходящей аорты и «бычьей дугой» ранее было показано наличие VUS в генах, связанных с наследственной аневризмой восходящей аорты FBN2, NOTCH1 и COL5A2 [3], возможно, что варианты с неопределенной клинической значимостью в данной категории генов влияют на развитие спорадической аневризмы в сочетании с «бычьей дугой».
Миссенс-вариант c.1151T>C (p.Ile384Thr) гена ACVR2B , выявленный в гетерозиготном состоянии у пациента с дилатацией аорты без аномалии строения дуги, ранее не был описан в литературе. Вместе с тем данный вариант находится в одном цитоплазматическом топологическом белковом домене с вероятно патогенным мис-сенс-вариантом p.Arg383Cys (rs1559655653), связанным с нарушением латеральности [21]. Белок, кодируемый геном ACVR2B , является рецептором активина А типа IIB – фактора роста и дифференцировки клеток, принадлежащего к суперсемейству сигнальных белков TGF-бета.
Заключение
Таким образом, редкие клинические значимые варианты генов эмбрионального развития сердечно-сосудистой системы могут быть связаны с развитием спорадической формы аневризмы восходящей аорты, где в сочетании с «бычьей дугой» аорты играют роль гены, регулирующие функционирование ресничек и микротрубочек CCDC39 и ANKS6. Патогенный вариант c.610-2A>G гена CCDC39 локализован в сайте сплайсинга, а VUS c.2564T>C в гене ANKS6 представляет собой миссенс-за-мену (p.Phe855Ser). Клинически значимые варианты, выявленные у больных с аневризмой восходящей аорты и «бычьей дугой», локализованы в сайтах связывания ТФ MEIS3, PKNOX1 и ElF5, необходимых для развития и функционирования сердечно-сосудистой системы. Поскольку нарушение нормального функционирования первичных ресничек является фактором развития различных врожденных аномалий, в том числе сердечно-сосудистой системы, вероятно, что определенный «груз» мутаций в генах цилиопатий может вносить вклад с формирование фенотипа аневризмы восходящей аорты, особенно в сочетании с аномалиями ее дуги.
