Инсулин, головной мозг, болезнь Альцгеймера: новые данные

Автор: Булгакова Светлана Викторовна, Романчук Петр Иванович, Тренева Екатерина Вячеславовна
Журнал: Бюллетень науки и практики @bulletennauki
Рубрика: Медицинские науки
Статья в выпуске: 3 т.6, 2020 года.
Бесплатный доступ
Последние четыре десятилетия ознаменовались рядом научных открытий. Так стало известно, что инсулин, рецепторы к нему найдены в структурах головного мозга. Кроме того, стала известна роль этого гормона в активации нейрональных стволовых клеток, росте, развитии нейрональной сети, синаптической передаче, когнитивных функций и так далее. Дисфункция передачи сигналов и метаболизма инсулина способствует развитию ряда дегенеративных заболеваний головного мозга. Все больше данных говорит о связи сахарного диабета 2 типа и болезни Альцгеймера, имеющих много общих патофизиологических характеристик. Данный обзор литература посвящен анализу клинических и экспериментальных данных, связывающих инсулин, инсулинорезистентность с дегенеративными процессами в головном мозге, оценке фармакологических стратегий, направленных на коррекцию сигнальных путей инсулина в ЦНС и когнитивных функций. Искусственный интеллект, нейросети «мозг-микробиота» позволяют управлять взаимодействием генетических и эпигенетических программ старения и здорового долголетия. Новая управляемая здоровая биомикробиота и персонализированное функциональное и сбалансированное питание «мозга и микробиоты» - это долговременная медицинская программа пациента, которая позволяет комбинированному применению питательной эпигенетики и фармэпигенетики, а главное проведению профилактики полипрагмазии.
Инсулин, рецепторы инсулина, болезнь альцгеймера, инсулиноподобный пептид 1, нейродегенеративные заболевания
Короткий адрес: https://sciup.org/14116201
IDR: 14116201 | УДК: 616.83/.85:616.89 | DOI: 10.33619/2414-2948/52/10
Insulin, brain, Alzheimer's disease: new evidence
The last four decades have been marked by a number of scientific discoveries. So, it became known that insulin, receptors for it are found in the structures of the brain. In addition, the role of this hormone in the activation of neuronal stem cells, growth, development of the neuronal network, synaptic transmission, cognitive functions and so on has become known. Signal dysfunction and insulin metabolism contribute to the development of a number of degenerative diseases of the brain. More and more evidence suggest a relationship between type 2 diabetes mellitus and Alzheimer’s disease, which share many common pathophysiological characteristics. This review of the literature is devoted to the analysis of clinical and experimental data linking insulin, insulin resistance with degenerative processes in the brain, and the evaluation of pharmacological strategies aimed at the correction of insulin signaling pathways in the central nervous system and cognitive functions. Artificial intelligence, brain-microbiota neural networks allow to control interaction of genetic and epigenetic programs of ageing and healthy longevity. The new managed healthy biomicrobiota and personalized functional and balanced nutrition of ‘brain and microbiota’ is a long-term medical program of the patient, which allows the combined application of nutritional epigenetics and pharmacepigenetics, and the main thing to carry out prevention of polypragmasia.
Текст обзорной статьи Инсулин, головной мозг, болезнь Альцгеймера: новые данные
Бюллетень науки и практики / Bulletin of Science and Practice
УДК 616.83/.85:616.89
Нейроось «микробиота–кишечник–мозг» представляет собой динамическую матрицу тканей и органов, включая желудочно–кишечную микробиоту, иммунные клетки, ткани кишечника, железы, вегетативную нервную систему и головной мозг, которые взаимодействуют сложным разнонаправленным образом через ряд анатомически и физиологически различных систем. Долгосрочные возмущения этой гомеостатической среды могут способствовать прогрессированию ряда нарушений путем изменения физиологических процессов, включая активацию гипоталамо–гипофизарно–надпочечниковой оси, нейромедиаторных систем, иммунной функции и воспалительной реакции [1].
Современные инструменты и методики эпигенетической, диетической и биомикробиотической защиты здорового старения — это междисциплинарные, межвузовские и межведомственные направления, которые фокусируются на изучении нервной системы и влияния мозга на поведение и мыслительную способность людей [1–2].
Новая эпигенетика Homo sapiens управляет взаимодействием эпигенетических механизмов старения и долголетия с биологией, биофизикой, физиологией и факторами окружающей среды в регуляции транскрипции. Старение — это структурно–функциональная перестройка (перепрограммирование) и постепенное снижение физиологических функций организма, которые приводят к возрастной потере профессиональной пригодности, болезням, и к смерти. Понимание причин здорового старения составляет одно из самых проблемных междисциплинарных направлений [2].
Генетический и эпигенетический вклад в старение и долголетие человека огромен. В то время как факторы окружающей среды и образа жизни важны в более молодом возрасте, вклад генетики проявляется более доминантно в достижении долголетия и здоровой старости. Эпигеномные изменения во время старения глубоко влияют на клеточную функцию и стрессоустойчивость. Дисрегуляция транскрипционных и хроматиновых сетей, вероятно, является важнейшим компонентом старения. В ближайшем будущем искусственный интеллект и крупномасштабная биоинформационная система анализа сможет выявить вовлеченность многочисленных сетей взаимодействия [2].
Раньше считалось, что мозг является нечувствительным к инсулину и неподверженным его влиянию органом, т. к. гормон не может проходить через гематоэнцефалический барьер (ГЭБ) [3]. Также отрицалась и вероятность локального синтеза инсулина в каком-либо отделе головного мозга. Однако в 1967 г. Р. Марголис и Н. Альтшулер доказали, что уровень инсулина повышается в цереброспинальной жидкости собак при его внутривенном введении. В связи с чем появилась версия о том, что гормон все же может пересекать ГЭБ через высоко специализированную транспортную систему [3–6].
Спустя 10 лет Я. Хавранкова и коллеги обнаружили сам инсулин и его рецепторы в разных отделах головного мозга крысы [6–7].
В настоящее время известно, что инсулинтранспортная система в различных областях мозга существенно различается, что приводит к дифференциации проницаемости инсулина для различных популяций нейронов, вследствие чего гипоталамус, продолговатый мозг, варолиев мост имеют более высокую концентрацию инсулина, а затылочная доля и таламус — сравнительно низкую [3, 8]. Инсулинтранспортная система существенно меняется в условиях голодания, переедания, при ожирении и старении, у пациентов с СД 2-го типа и болезнью Альцгеймера (БА). Кроме того, существует и вторая версия появления инсулина в головном мозге — синтез гормона непосредственно в головном мозге. Эти представления базируются на обнаружении мРНК для инсулина в гипоталамусе, гиппокампе и культурах нейронов [6, 9].
Тем не менее, в настоящее время широко признано, что инсулин играет важную роль в жизнеспособности нейронов и функционировании головного мозга. Фактически, действие инсулина необходимо для синаптической пластичности нейронов и способствует обучению и памяти [10] . Также было показано, что инсулин способствует образованию нейронной сети, активации нейрональных стволовых клеток, росту, репарации и нейропротекции нейронов, регуляции энергетического обмена, защите клеток от окислительного стресса [11] .
Следовательно, изменения в метаболизме и передаче сигналов инсулина в центральной нервной системе (ЦНС) могут способствовать развитию ряда заболеваний головного мозга.
За последние 20 лет многие исследования показали связь между нейродегенеративными расстройствами, такими как БА и нарушением передачи сигналов инсулина в ЦНС [12– 13], предполагая, что снижение действия инсулина и инсулинорезистентность могут играть важную роль в патогенезе этих заболеваний.
Инсулин и инсулиновая сигнальная система в головном мозге
Инсулин и инсулиноподобный фактор роста (IGF)-1 регулируют ряд биологических процессов посредством связывания и активации двух близкородственных рецепторов тирозинкиназы, рецептора инсулина (IR) и рецептора IGF-1 (IGF-1R) [14]. Несколько исследований показали, что IR и IGF-1R, а также их общие нижестоящие пути в большом количестве находятся в головном мозге, и, что более важно, эти пути функционируют как регуляторы нейрогенеза, функций мозга и энергетического баланса и системного гомеостаза [12]. Наибольшая концентрация IR находится в гипоталамусе, гиппокампе, в обонятельной луковице, мозжечке, миндалине и коре головного мозга [15], что свидетельствует о многофункциональности инсулина [12].
Инсулин — это пептидный гормон, состоящий из двух цепей и 51 аминокислотного остатка, не может пассивно проходить через гематоэнцефалический барьер (ГЭБ), но, тем не менее, он обнаружен в спинномозговой жидкости (СМЖ). Происхождение «мозгового» инсулина является спорным. Одна из гипотез заключается в том, что плазменный инсулин способен проникать через ГЭБ через насыщаемый транспортный процесс, возможно, через 1R-сосудистого эндотелия. Подтверждением этой гипотезы является доказательство того, что уровни инсулина в СМЖ ниже (примерно на 25%) циркулирующих в крови, и его концентрации увеличиваются после еды или при периферической инфузии инсулина [16]. Другой возможностью является доказательство того, что существуют области мозга, такие как гипоталамус, в которых отсутствует эффективный барьер, обеспечивающий доступ инсулина к ЦНС [17]. Третья гипотеза предполагает, что инсулин синтезируется в областях мозга, но это предположение требует дальнейших исследований [12, 18].
После достижения ЦНС инсулин связывается с IR, который принадлежит к семейству рецепторов тирозинкиназы. Интересно, что IR-субъединицы, обнаруженные в головном мозге, имеют структуру, отличную от периферических, и основным отличием является более низкая молекулярная масса IR-субъединиц мозга, вероятно, из-за различного гликозилирования [19]. Более того, мозг экспрессирует преимущественно изоформу A (- exon 11) IR, которая имеет более высокое сродство к IGF-2, в отличие от периферических тканей, которые преимущественно экспрессируют изоформу B (+ exon 11) [20–21] ,
Предполагается, что инсулин обладает нейропротекторными свойствами и оказывает нейротрофическое действие на нейроны ЦНС [22]. Более того, это может положительно влиять на когнитивные функции, включая эмоции, внимание, исполнительное функционирование, обучение и память [23].
После связывания инсулина с IR происходит аутофосфорилирование рецептора, и активированный IR фосфорилирует каскад белков субстрата IR (Рисунок) [14].
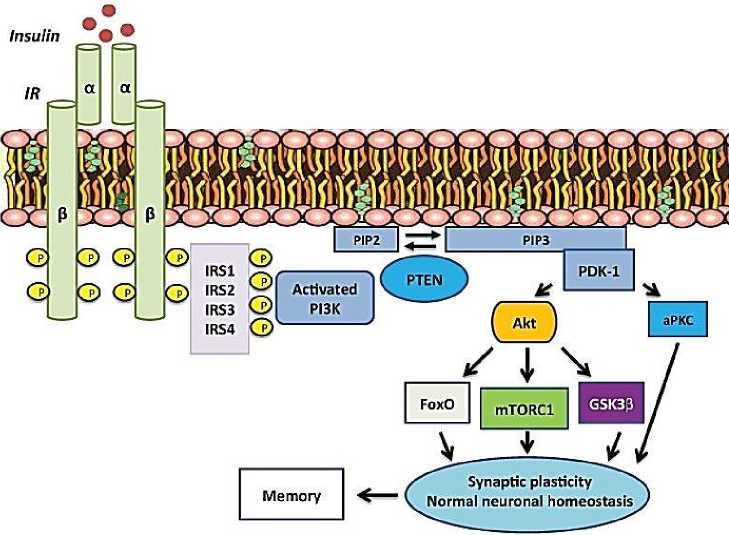
Рисунок. Инсулиновый сигнальный путь [6].
Среди IR-субстратов (IRS) мРНК IRS-2 в головном мозге является наиболее распространенной по сравнению с IRS-1; IRS-4, которые, в основном, экспрессируются в эмбриональном развитии, также в мозге взрослых мышей, особенно в гипоталамусе [12, 24]. На уровне всего тела IRS-1, по-видимому, является критическим для роста, а мыши с IRS-1-нулем приводят к увеличению соотношения мозг–тело [25]. С другой стороны, разрушение гена IRS-2 снижает пролиферацию нейронов во время развития на 50%, и, как следствие, у мышей с нулевым IRS-2 наблюдается пониженное отношение поверхности головного мозга к поверхности тела [26]. Кроме того, во время старения нейрофибриллярные клубки, содержащие фосфорилированный тау, накапливались в гиппокампе у мышей, нокаутированных по IRS-2, предполагая, что передача сигналов IRS-2 является нейрозащитной [26]. Несмотря на это, IRS-2-нулевые мыши являются долгоживущими [27], что согласуется с ролью центральной передачи сигналов инсулин/IGF в контроле продолжительности жизни у млекопитающих. IRS-4 может синергетически взаимодействовать с IRS-2 в гипоталамусе для контроля потребления пищи, расхода энергии и метаболизма глюкозы [28].
После связывания инсулина с рецептором инсулина (IR) происходит аутофосфорилирование, которое необходимо для его активации. Затем активированный рецептор инсулина фосфорилирует белки IRS. IRS активируют PI3K, который катализирует добавление фосфатной группы к мембранному липиду PIP2, тем самым превращая его в PIP3. PTEN может преобразовать PIP3 обратно в PIP2. Связанный с мембраной PIP3 рекрутирует и активирует PDK-1, который фосфорилирует и активирует Akt и атипичные PKC. Akt опосредует большинство метаболических эффектов инсулина и синаптической пластичности мозга, нейронального гомеостаза и памяти (рисунок 1) [6].
Сокращения: IRS (субстрат рецептора инсулина), PI3K (фосфатидилинозитол-3-киназа), PIP2 (фосфатидилинозитол-4,5-бисфосфат), PIP3 (фосфатидилинозитол-3,4,5-трифосфат), PTEN (гомолог фосфатазы и тензина).
Специфическая инактивация IR в головном мозге (то есть нейрон-специфических IR-нокаутов — NIRKO-мышей) показала, что недостаток IR в мозге определяет измененные метаболические фенотипы, включая ожирение, инсулинорезистентность, играет важную роль в регуляции энергетического метаболизма [12, 29]. Более того, инсулинорезистентность в головном мозге существует как явление, независимое от периферической инсулинорезистентности и углеводного обмена [30–31]. Это означает, что сниженная чувствительность к инсулину в головном мозге имеет иные последствия, чем в периферических тканях. Недавно опубликованные данные показали, что периферическая толерантность к инсулину и глюкозе была сопоставимой у пожилых мышей дикого типа и мышей APP / PS1 (модель БА), в то время как уровни серинового фосфорилированного IRS-1 были повышены в мозге мышей APP/PS1 [32]. Это дает некоторую поддержку предположению о том, что центральная инсулинорезистентность может существовать самостоятельно отдельно от периферической и связана с дегенеративными процессами при БА.
Одним из основных нижестоящих путей белков IRS является каскад PI3K / Akt. Это, в свою очередь, нацелено на множественные нисходящие пути, включая mTORC1, гликогенсинтазкиназу 3β (GSK-3β) и семейство транскрипционных факторов FoxO (Figure 1) [33]. Было показано, что многие из этих путей играют ключевую роль в нормальной работе головного мозга.
СД2 и нейродегенеративные процессы: роль нарушений в инсулиновой сигнальной системе головного мозга, инсулинорезистентности и гиперинсулинемии
СД2 представляет собой хроническое заболевание, связанное с возрастом, с растущей распространенностью. В настоящее время во всем мире более четырехсот миллионов человек страдают сахарным диабетом, и ожидается, что это число резко возрастет в течение следующих тридцати лет [34]. СД2, характеризующийся инсулинорезистентностью и хроническим воспалением, вызывает ускоренное старение [35] и приводит к преждевременной заболеваемости и смертности. Влияние СД2 на головной мозг в настоящее время хорошо известно: заболевание является основным фактором риска снижения когнитивных функций и деменции. Фактически, СД2 увеличивает долгосрочный риск развития деменции почти в 2 раза, и каждый десятый случай деменции среди населения мира может быть связан с последствиями СД2 [13]. Взаимная связь в распространенности этих хронических заболеваний обусловлена тем, что диабет и деменция имеют несколько общих особенностей, приводящих к повреждению головного мозга, наиболее важными из которых являются нарушение чувствительности к инсулину, накопление бета-амилоида (Aβ), гиперфосфорилирование тау, повреждение сосудов и воспаление.
БА является хроническим нейродегенеративным заболеванием, которое обычно начинается медленно и ухудшается со временем. Это является причиной 60–70% случаев деменции [36]. Наиболее распространенным ранним симптомом является трудность запоминания недавних событий (кратковременная потеря памяти). По мере развития болезни симптомы могут включать проблемы с речью, дезориентацию, перепады настроения, потерю мотивации, неспособность к самообслуживанию и поведенческие проблемы [37]. Это прогрессирующее нейродегенеративное заболевание характеризуется накоплением в мозге внеклеточных нейритных бляшек и фибрилл (в основном состоящих из агрегированных амилоидных β-Aβ-пептидов), внутриклеточных нейрофибриллярных клубков (накопление гиперфосфорилированных белков tau-NFTs), микроглиальной инфильтрации, атрофия головного мозга и широко распространенная потеря синаптической и нейрональной передачи. Выраженное нейровоспаление также постоянно наблюдается при БА [38]. Данные исследований показали, что гиперактивность провоспалительных маркеров в головном мозге предшествует развитию бляшек и нейрофибриллярных клубков при БА [39].
Обнаружено ряд патофизиологических связей между БА и нарушениями обмена веществ, такими как СД2, ожирение и метаболический синдром [40–42]. В отличие от небольшого количества случаев БА (~ 3%), обусловленных наследственными генетическими причинами, патогенез и этиология спорадической БА с поздним началом являются многофакторными, включающими генетические факторы и факторы образа жизни [43]. Признание СД2 в качестве основного фактора риска развития деменции, особенно БА, побудило исследователей к поиску основных механизмов, связывающих эти два возрастных хронических заболевания. Известно, что метаболические нарушения, характерные для СД2 (например, гипергликемия, гиперинсулинемия, гиперхолестеринемия), связаны с атрофией головного мозга и патологическими признаками БА [14, 44]. Является ли резистентность к инсулину причиной или следствием БА, пока неясно.
Более того, ряд исследователей подтверждают версию о том, что нарушение регуляции инсулиновой сигнальной системы может быть ключевым фактором, способствующим раннему развитию БА. Так, некоторые ученые показали, что экспрессия и активация белков IR, IGF-1R и IRS-1 снижена в мозге пациентов с БА по сравнению с контрольной группой [45]. Более того, некоторые авторы продемонстрировали, что неокортикальные уровни инсулина и связывание с IR снижаются в мозге больных БА [46]. Наконец, более низкая концентрация инсулина в СМЖ, несмотря на более высокую концентрацию инсулина в плазме [47], предполагает снижение действия инсулина в ЦНС.
Амилоидные бляшки, обнаруженные в мозге пациентов с БА, в основном состоят из Аβ, пептида, происходящего из более крупной молекулы, известной как белок-предшественник амилоида (АРР). Дисбаланс между продукцией, клиренсом и агрегацией пептидов вызывает накопление Aβ, и этот избыток может быть инициирующим фактором для развития БА [48].
Ряд исследователей предположили связь между дефектами энергетического метаболизма и функциональными изменениями, связанными с развитием БА [49]. Ингибирование энергетического обмена может изменить процесс APP и вызвать продукцию амилоидогенных продуктов [50]. Связь между инсулином и метаболизмом Aβ в последнее время привлекает все большее внимание ученых [51].
Известно, что малые олигомеры Aβ способствуют синаптотоксичности и последующим изменениям, которые приводят к нейродегенеративным процессам при БА [52–53]. Как часть этих нейродегенеративных процессов, олигомеры Aβ, по-видимому, оказывают негативное влияние на передачу сигналов инсулина, ингибируя аутофосфорилирование рецептора [54], и заметно снижают уровни IR и их активность на клеточной поверхности дендритов нейронов гиппокампа [55]. IR играют ключевую роль в важных неврологических процессах, включая обучение и память, а также фосфорилирование тау. Таким образом, индуцированная олигомерами Aβ потеря мембранных IR может представлять собой важный ранний механизм, лежащий в основе нарушения памяти и других патологических нарушений при БА.
Также было обнаружено, что олигомеры Aβ определяют аберрантную активацию ингибирования TNFα / JNK и IRS-1 как в моделях in vitro, так и in vivo [30, 56]. Кроме того, олигомеры Aβ также оказывают влияние на передачу сигналов ниже IRS-1 и PI3K, где они могут активировать сериновое фосфорилирование Akt и стимулировать воспалительные процессы [57]. С другой стороны, известно, что резистентность к инсулину ускоряет выработку Aβ, способствуя его накоплению. Когда резистентность к инсулину индуцируется у трансгенных мышей с БА или у мышей с диабетом, страдающих ожирением, путем их кормления пищей с высоким содержанием жира, мыши демонстрируют повышенные уровни Aβ в мозге и рост уровней ключевых ферментов, которые генерируют Aβ (например, γ-секретазу) [58]. Наконец, инсулин и Aβ являются субстратами инсулин-разлагающего фермента, и было высказано предположение, что гиперинсулинемия ингибирует деградацию Aβ путем конкурентной блокировки инсулин-разлагающего фермента [59].
Нарушение в инсулиновой сигнальной системе головного мозга и гиперфосфорилирования тау
Дефицит передачи сигналов инсулина также может усугублять нейродегенерацию за счет увеличения фосфорилирования тау-белка, являющимся нейрональным микротрубочковым белком, обнаруженным в аксонах. Он играет важную роль в сборке и стабильности микротрубочек, а также в транспорте везикул в нейронах. При БА гиперфосфорилирование тау белка является важным патологическим признаком, и способствуют дисфункции и дегенерации нейронов [60]. Было показано, что инсулин и IGF-1 регулируют фосфорилирование тау путем ингибирования GSK-3β в нейронах культуры клеток [61]. GSK-3β представляет собой ключевую киназу, которая фосфорилирует тау белок. Недостатки или нарушения в передаче сигналов инсулина в головном мозге приводят к снижению активности Akt, что ведет к увеличению активности GSK-3β. Это явление вызывает гиперфосфорилирование тау и, следовательно, образование тау-фибрилл [62]. Более того, периферическая гиперинсулинемия способствует фосфорилированию тау in vivo [63] . Было продемонстрировано, что при делеции гена IGF-1 и IRS-2 фосфорилирование тау резко увеличивается у мышей, нокаутированных по IGF-1 и IRS-2 [26, 64]. Действительно, генетическая делеция IGF-1 специфически увеличивает фосфорилирование тау в двух локусах-мишенях GSK-3β [57]. Эти результаты предполагают, что нормальная передача сигналов инсулина и IGF-1 предотвращает гиперфосфорилирование тау в мозге. Учитывая, что СД2 характеризуется инсулинорезистентностью, гиперинсулинемией и нарушением передачи сигналов инсулина, неудивительно, что повышенная активность GSK-3β при СД2 может привести к увеличению продукции Аβ [65] и повышенному фосфорилированию тау [66].
Инсулин также может регулировать экспрессию тау, и снижение передачи сигналов инсулина может привести к нарушению экспрессии гена тау [57], что приводит к снижению уровня нормального растворимого тау, в то время как гиперфосфорилированный тау накапливается, усугубляя коллапс нейронального цитоскелета, ретракцию нейритов и нарушения в образовании синапсов. Более того, было продемонстрировано, что ассоциированное с БА снижение экспрессии мРНК tau коррелирует с нарушением передачи сигналов инсулина и IGF-1, наблюдаемым в тех же самых образцах БА [67], демонстрируя сильную связь между этими двумя механизмами.
Инсулинорезистентность, васкулопатия головного мозга и нейровоспаление
Нейродегенеративные расстройства и СД2 характеризуются как сосудистым поражением, снижением мозгового кровотока, так и аберрантным воспалительным ответом [68].
Было доказано, что у пациентов с БА наблюдается снижение регионарного мозгового кровотока, что может привести к снижению снабжения мозга кислородом, глюкозой и питательными веществами [68–69]. Это явление связано с нарушением пути трансдукции инсулина. Инсулиновая сигнальная система участвует в регуляции вазодилатации и вазоконстрикции [70]. Активация IR опосредует вазодилатацию через путь PI3K / Akt. Он стимулирует эндотелиальную синтазу оксида азота (eNOS), что приводит к выработке оксида азота (NO) и сосудистой релаксации [70]. В инсулинорезистентном состоянии наблюдается специфическое нарушение вазодилататорного пути PI3K, что приводит к снижению продукции NO и, следовательно, к вазоконстрикции. В результате происходит снижение поступления питательных веществ в головной мозг, увеличение окислительного стресса и продукции активных форм кислорода (АФК) и, следовательно, активация реакции воспаления. Выброс провоспалительных цитокинов и рекрутирование макрофагов провоцируют атеросклероз, что в конечном итоге приводит к макрососудистым осложнениям [68].
Хорошо известно, что процессы хронического воспаления составляют основную часть патогенеза СД2, а также нейродегенеративных заболеваний. Ряд исследователей доказали, что индуцированное хроническое воспаление является важной ранней стадией патогенеза БА [71–72].
Было показано, что гиперинсулинемия способствует развитию процессов воспаления в ЦНС [73]. Установлено, что повышение уровня периферического инсулина приводит к увеличению в головном мозге уровней провоспалительных цитокинов, таких как интерлейкин-1 (IL-1), интерлейкин-6 (IL-6) и фактор некроза опухоли-α (TNF-α), повышенных при БА и локализованных в амилоидных бляшках и связанных с ними глиальных клетках [74].
При периферической инсулинорезистентности выработка воспалительных цитокинов и активация передачи сигналов о воспалительном стрессе могут привести к сериновому фосфорилированию IRS-1 с помощью киназ, ингибитора каппа-B-киназы (IKK), c-Jun N-терминальной киназы (JNK) и ERK2, которая, в свою очередь, нарушает IR-опосредованную передачу сигналов, блокируя внутриклеточное действие инсулина [75]. Предполагается, что подобный механизм встречается в головном мозге, где олигомеры Aβ могут активировать микроглию, что приводит к секреции провоспалительных цитокинов, которые связываются с их соответствующими рецепторами, активируя одну или несколько серинкиназ IRS-1 и, в свою очередь, фосфорилируя IRS [56]. Повышенные уровни сосудистых провоспалительных цитокинов, наблюдаемые как при СД2, так и при БА, также могут влиять на передачу сигналов инсулина в головном мозге. При повреждении ткани сосудов головного мозга цитокины могут пересекать ГЭБ и активировать фосфорилирование IRS-1 [76–77].
Сосудистое воспаление также может быть опосредовано активацией и увеличением количества рецепторов конечных продуктов гликирования (RAGE). RAGE экспрессируется в нейрональных клетках, астроцитах микроглии и в эндотелиальных клетках головного мозга, и уровни его повышаются как при БА, так и при СД 2. Повышенные уровни RAGE были предложены в качестве возможного механизма сосудистой дисфункции как при СД2, так и при БА [78], а взаимодействие между нарушенным церебральным метаболизмом глюкозы, окислительным стрессом и накоплением конечных продуктов гликирования играет важную роль в порочном цикле, который способствует прогрессированию БА [79]. RAGE представляет собой путь опосредованного рецептором транспорта Aβ через ГЭБ от периферии к мозгу [80], индуцируя цереброваскулярную дисфункцию, приводящую к нервно-сосудистому стрессу, выработке TNF-α и IL-6, способствуя синаптотоксичности и нейродегенерации [78].
Лекарственная коррекция нейродегенеративных изменений головного мозга с учетом патогенеза
Поскольку СД2 имеет несколько общих патогенетических характеристик с нейродегенеративными расстройствами, как обсуждалось ранее, было высказано предположение, что некоторые препараты, используемые при терапии СД2, могут иметь потенциальную пользу при лечении БА:короткая характеристика препаратов представлена ниже:
Метформин
Восстанавливает митохондрии, ослабляет эффекты AGE путем активации AMPK в нейронах [81–82].
Активирует передачу сигналов инсулина и уменьшает фосфорилирование тау в клеточных линиях нейронов [83].
Индуцирует протеинфосфатазу 2А и снижает фосфорилирование тау в нейронах трансгенной мыши Тау [84].
Ослабляет когнитивные нарушения у мышей с ожирением, устойчивых к лептину [85].
Увеличивает выработку бета-белка амилоида в клеточных моделях человека ( отрицательный эффект ) [86].
Уменьшает риск снижения когнитивных функций у больных диабетом [87].
Улучшает когнитивные функции у пациентов с депрессией [88–89].
Увеличивает риск когнитивных нарушений в исследованиях, проведенных на пациентах с БА ( отрицательный эффект ) [90].
Препараты сульфонилмочевины
Глимепирид защищает нейроны от индуцированной бета-амилоидом дегенерации синапсов in vitro [91].
Гликлазид оказывает антиоксидантное действие на мозг, у крыс с диабетом [92] Глибенкламид уменьшает депрессию и беспокойство у крыс с БА [93]
В сочетании с метформином, снижают риск развития деменции у пациентов с диабетом [94].
Глитазоны
Нейропротективные эффекты при БА, связанные с ингибированием воспаления и отложения Aβ [95].
Пиоглитазон препятствует снижению глиальной активации у мышей с БА [96]
Пиоглитазон усиливает передачу сигналов Akt и гиперфосфорилирование тау у мышей с БА [97].
В сочетании с лептином пиоглитазон снижает уровень амилоида в мозге у мышей с БА [98].
Пиоглитазон улучшает когнитивные функции и регионарный мозговой кровоток у пациентов с СД 2 [99].
Пиоглитазон может обеспечить улучшение когнитивных функций на ранних стадиях и при легких и умеренных проявлениях БА у людей [100].
Агонисты глюкагоноподобного пептида 1
Уменьшают окислительный стресс и апоптоз клеток головного мозга; улучшает синаптическую пластичность у мышей с БА [101].
Влияют на клеточные механизмы нейрональной защиты и митохондриальной функции [102].
Снижение фосфорилирования тау, предотвращение синаптической потери, уменьшение отложения Aβ у мышей с БА [103–104].
Предотвращают снижение метаболизма глюкозы в головном мозге у пациентов с БА [105].
Ингибиторы ДПП-4
Снижение фосфорилирования тау, амилоидной нагрузки и когнитивных нарушений с улучшением памяти [106–107].
Улучшение уровня инкретина, уменьшение отложения Aβ, фосфорилирования тау, активации GSK-3β и АФК [108].
Улучшение контроля глюкозы и предотвращение ухудшения когнитивных функций у пожилых пациентов с СД2 [109].
Инсулин
Ослабляет когнитивные нарушения и улучшает память у взрослых с БА [110–111].
In vitro подавляет апоптоз; in vivo регулирует фосфорилирование тау, метаболизм и клиренс Aβ [112].
Улучшает память, настроение, церебральный метаболизм глюкозы; сохраняет объем мозга у пациентов с БА [113].
Метформин
Метформин — бигуанид, снижает опосредованную инсулином выработку глюкозы в печени, повышает чувствительность к инсулину и представляет собой терапию первой линии при СД2. Он быстро пересекает ГЭБ, распределяется по областям головного мозга [114] и, благодаря активации пути AMPK, по-видимому, оказывает нейропротективное действие на нервные стволовые клетки человека, восстанавливая функции митохондрий и ослабляет эффекты конечных продуктов гликирования [81–82].
Данные о влиянии метформина на нейродегенеративные нарушения противоречивы. В исследованиях in vitro сообщалось о способности метформина снижать фосфорилирование тау в клеточных линиях нейронов [83–84]. Исследования in vivo показали, что у мышей с ожирением, устойчивых к лептину, метформин ослаблял когнитивные нарушения и БА-подобную патологию [85]. Напротив, исследование культуры клеток показало, что метформин увеличивает выработку Aβ [86].
Наблюдательные исследования у лиц с СД2, принимающих метформин, показывают снижение проявлений легкой когнитивной недостаточности (MCI) [87] и деменции [94, 115] по сравнению с плацебо. Длительное лечение метформином, по-видимому, уменьшает риск снижения когнитивных функций у пациентов с диабетом [85] и снижает депрессивные и улучшает когнитивные функции, изменяя метаболизм глюкозы, у пациентов с депрессией [88]. Пилотное клиническое исследование пациентов с MCI в течение 12 месяцев показало, что метформин улучшал когнитивные функции у людей без диабета по сравнению с плацебо [116].
Клиническое исследование, в котором изучалось влияние различных методов лечения СД2 на когнитивные функции, показало, что пациенты с диабетом, которые использовали только метформин, обладали лучшими когнитивными функциями в области словесного обучения, рабочей памяти и исполнительной функции по сравнению с участниками других форм лечения диабета [89].
С другой стороны, повышенный риск когнитивных нарушений и развития БА был продемонстрирован с использованием метформина в исследовании, проведенном на пациентах с БА [90]. Это явление было частично обусловлено дефицитом витамина В12, вызванным метформином. Тем не менее, результаты анализа когнитивных функций, проведенный через 8–10 лет после терапии метформином в рамках исследования результатов программы профилактики диабета (DPPOS) [117], не показали какого-либо негативного влияния от длительного применения метформина.
Планируемые в настоящее время рандомизированные клинические исследования позволят оценить, может ли метформин предотвратить снижение когнитивных функций или улучшить когнитивные функции у людей [118].
Препараты сульфонилмочевины
Препараты сульфонилмочевины — это сахароснижающие препараты, которые стимулируют высвобождение инсулина, блокируя чувствительные к АТФ калиевые каналы бета-клеток поджелудочной железы. In vitro глимепирид защищает нейроны от бета-амилоид-индуцированной дегенерации синапсов [91]. У крыс с диабетом, индуцированным стрептозотоцином, гликлазид оказывал антиоксидантное действие на головной мозг [85]. Кроме того, глибенкламид снижает депрессию и тревожность у крыс с БА [93].
Клиническое проспективное исследование, проведенное в течение 8 лет на пациентах с СД2, показало, что комбинация препаратов сульфонилмочевины и метформина снижала риск развития деменции [94], однако, другое исследование «случай–контроль» показало, что длительное использование препаратов сульфонилмочевины не влияет на риск развития деменции [119].
Для подтверждения потенциальной терапевтической роли этого класса лекарств необходимы дальнейшие исследования.
Тиазолидиндионы (глитазоны)
Тиазолидиндионы (TZD) (пиоглитазон и росиглитазон) являются мощным и селективным стимулятором ядерных гамма-рецепторов, активируемых пролифератором пероксисом (гамма-PPAR), которые улучшают чувствительность к инсулину в мышечной, жировой и печеночной тканях; снижают системную инсулинорезистентность. TZD могут играть роль в улучшении функции нейронов и формировании памяти. Эти препараты показали нейропротекторные эффекты при БА, связанные с ингибированием экспрессии воспалительных генов и изменением образования и отложения Aβ [95].
Пиоглитазон способен проникать в головной мозг, подавляет глиальную активацию и уменьшает клинические проявления БА [96] .
У мышей с БА пиоглитазон, вводимый в течение 4 месяцев, усиливает передачу сигналов Akt, улучшает пространственное обучение и снижает гиперфосфорилирование тау [95] . Кроме того, применение в комбинации с лептином уменьшает дефицит памяти и уровень амилоида в головном мозге [98].
Пилотное исследование пациентов с СД2, получавших пиоглитазон в течение 6 месяцев, показало улучшение когнитивных функций и регионарного мозгового кровотока в теменной доле [99]. Однако 18-месячное исследование пациентов с БА без диабета, направлененое на оценку безопасности пиоглитазона, показало отсутствие влияния на когнитивные функции [120].
Мета-анализ влияния PPAR-гамма-агонистов у пациентов с БА показал, что только пиоглитазон может обеспечить клиническое улучшение на ранних стадиях БА от легкой до умеренной степени [100, 122].
Фаза 3 клинического испытания эффективности пиоглитазона у пациентов с легкими когнитивными нарушениями с использованием алгоритма для оценки генетических биомаркеров для доклинической диагностики, таких как статус APOE и генотипы продолжается. (Идентификатор клинического испытания NCT01931566) . Данные будут доступны в 2020 году.
Агонисты рецепторов глюкагоноподобного пептида-1 (GLP-1)
Другой класс сахароснижающих препаратов — агонисты рецептора GLP-1. GLP-1 представляет собой инкретиновый пептид, секретируемый кишечником, который усиливает глюкозозависимую секрецию инсулина и ингибирует секрецию глюкагона. GLP-1 также обладает трофическими свойствами, такими как стимуляция неогенеза, роста и дифференцировки β-клеток, ингибирование апоптоза β-клеток и повышение выживаемости клеток [122–123].
GLP-1 и большинство аналогов пересекают ГЭБ и рецептор GLP-1 экспрессируется во многих отделах головного мозга, таких, как лобная доля, гипоталамус, таламус, гиппокамп, мозжечок и черная субстанция [124]. GLP-1 играет нейропротекторную роль: в мозге мышей с БА, по-видимому, за счет снижения апоптоза, защиты нейронов от окислительного стресса и синапсов от вредного воздействия пониженной синаптической пластичности в гиппокампе, вызванной Aβ [101]. Нативный GLP-1 имеет короткий период полураспада, поскольку он легко разлагается дипептидилпептидазой-4 (DPP-4). Было разработано несколько более стабильных чем нативный аналогов GLP-1. Среди них эксенатид, лираглутид и ликсисенатид, которые проходят через ГЭБ и, независимо от их влияния на контроль глюкозы, влияют на клеточные пути нейрональной защиты, митохондриальной функции, апоптоза и окислительного стресса [102]. Из-за их нейропротекторных эффектов аналоги GLP-1 были изучены, как потенциальное препараты для лечения БА и других нейродегенеративных расстройств. В исследованиях на мышиной модели БА аналоги GLP-1 снижали гиперфосфорилирование тау нейронов, предотвращали синаптическую потерю, улучшали моторную функцию, улучшали синаптическую пластичность, ослабляли дефицит памяти и обучения и уменьшали количество Aβ в головном мозге [103–104].
Нейропротективные эффекты лираглутида, по-видимому, опосредованы через сигнальный путь PI3K-Akt [125], тогда как эффекты ликсисенатида были отнесены к индуцированным сигнальным путям Akt и MEK [126].
Пилотное клиническое исследование показало, что 6-месячное лечение пациентов с БА лираглутидом предотвращает снижение метаболизма глюкозы в мозге, что уменьшает риск прогрессирования заболевания [105]. Другие исследования, оценивающие эффективность у пациентов с БА аналогов GLP-1, продолжаются.
Разумеется, аналоги GLP-1 имеют то преимущество, что не влияют на уровень сахара в крови у людей, не страдающих диабетом, и поэтому могут представлять потенциальное безопасное лечение БА или других нейродегенеративных состояний также у пациентов без СД2.
Ингибиторы дипептидилпептидазы-4 (DPP-4)
DPP-4 являются сахароснижающими препаратами, которые, ингибируют DPP-4 — протеолитический фермент, ответственный за деградацию GLP-1, продлевают период его полужизни в плазме, стабилизируя его уровень и вызывая функциональное усиление его сахароснижающего эффекта. Ингибиторы DPP-4 показали нейропротективные эффекты, которые могут быть частично опосредованы эффектами GLP-1 в мозге.
На моделях животных с БА лечение ингибиторами DPP-4 (саксаглиптин, вилдаглиптин, ситаглиптин) снижает фосфорилирование тау, амилоидную нагрузку и маркеры воспаления, также устраняет когнитивный дефицит с улучшением памяти [106–107].
В нейрональных клетках человека линаглиптин снижает отложение Aβ, гиперфосфорилирование тау, предотвращает активацию GSK3β и ослабляет внутриклеточную продукцию ROS, стимулируя передачу сигнала 5’АМФ-активируемой протеинкиназы (AMPK) -Sirt1 [108]. Все эти эффекты способствуют улучшению когнитивных функций.
У пожилых пациентов, страдающих СД2 и умеренными когнитивными нарушениями, лечение ингибитором DPP-4 улучшает контроль глюкозы и предотвращает ухудшение когнитивных функций [109]. Также в проспективном клиническом исследовании, оценивающем 6-месячное лечение ситаглиптином у пожилых пациентов с СД2, сообщалось об улучшении когнитивной функции [127].
Инсулин
Инсулин оказывает несколько воздействий на мозг в отношении познания, обучения, памяти и синаптической пластичности, возможно, вовлекая сложный путь инсулиновой сигнальной системы головного мозга / IR. Введение инсулина замедляет снижение когнитивных функций [110, 128–129] и улучшает память у взрослых с БА [111]. Однако системное введение инсулина характеризуется низким проникновением в головной мозг и повышенным риском гипогликемии. По этим причинам в нескольких клинических исследованиях было проведено изучение интраназального введения инсулина. После интраназального введения инсулин, минуя ГЭБ, достигает биологически значимых концентраций в мозге [111]. In vitro инсулин ингибирует апоптоз нейронов посредством активации протеинкиназы B и in vivo регулирует фосфорилирование тау, метаболизм белка-предшественника Aβ и клиренс Aβ [112].
Интраназальное введение инсулина улучшает память и настроение у здоровых взрослых, а также у пациентов с умеренными когнитивными нарушениями и поздним началом БА, у которых улучшается церебральный метаболизм глюкозы и сохраняется объем областей мозга [113]. Терапевтическое воздействие инсулина на ЦНС зависит от его дозы и модулируется генотипом APOE, сильным генетическим предиктором развития БА [111].
Выводы
В настоящее время признано, что инсулин может оказывать важное влияние на работу головного мозга.
Изменения метаболизма и передачи сигналов инсулина могут способствовать развитию нейродегенеративных заболеваний, таких как БА.
Ряд исследований in vivo и in vitro подтверждают тесную связь между СД2 и нейродегенеративными процессами в головном мозге и предполагают потенциальную терапевтическую роль некоторых сахароснижающих препаратов в профилактике и лечении БА. Однако не полная изученность этих вопросов диктует необходимость дальнейших исследований.
Список литературы Инсулин, головной мозг, болезнь Альцгеймера: новые данные
- Романчук П. И. Возраст и микробиота: эпигенетическая и диетическая защита, эндотелиальная и сосудистая реабилитация, новая управляемая здоровая биомикробиота // Бюллетень науки и практики. 2020. Т. 6. №2. С. 67-110. DOI: 10.33619/2414-2948/51/07
- Романчук П. И., Волобуев А. Н. Современные инструменты и методики эпигенетической защиты здорового старения и долголетия Homo sapiens // Бюллетень науки и практики. 2020. Т. 6. №1. С. 43-70. DOI: 10.33619/2414-2948/50/06
- Сухов И. Б. Нарушения гормональной регуляции аденилатциклазной системы в мозге крыс с сахарным диабетом и их коррекция с помощью интраназально вводимых инсулина и серотонина: автореф. дис.. канд. биол. наук. Санкт-Петербург, 2016.
- Булгакова С. В., Романчук П. И., Волобуев А. Н. Нейросети: нейроэндокринология и болезнь Альцгеймера // Бюллетень науки и практики. 2019. Т. 5. №6. С. 112-128. DOI: 10.33619/2414-2948/43/16
- Булгакова С. В., Романчук П. И., Волобуев А. Н. Клинико-биофизические принципы лечения сосудистой деменции и болезни Альцгеймера // Бюллетень науки и практики. 2019. Т. 5. №5. С. 57-72. DOI: 10.33619/2414-2948/42/08
- Tumminia A. et al. Type 2 diabetes mellitus and Alzheimer's disease: role of insulin signalling and therapeutic implications // International journal of molecular sciences. 2018. V. 19. №11. P. 3306.
- DOI: 10.3390/ijms19113306
- Волобуев А. Н., Романчук П. И., Булгакова С. В. Нейросеть "мозг-микробиота": регуляция "висцерального" мозга и накопление когнитивной памяти // Бюллетень науки и практики. 2019. Т. 5. №2. С. 33-52.
- DOI: 10.33619/2414-2948/39/05
- Волобуев А. Н., Пятин В. Ф., Романчук Н. П., Булгакова С. В. Давыдкин И. Л. Когнитивная дисфункция при перевозбуждении структур головного мозга // ВРАЧ. 2018. T. 29. №9. С. 17-20.
- DOI: 10.29296/25877305-2018-09-04
- Волобуев А. Н., Романчук П. И., Романчук Н. П., Давыдкин И. Л., Булгакова С. В. Нарушение памяти при болезни Альцгеймера // ВРАЧ. 2019. T.30. №6. С. 10-13.
- DOI: 10.29296/25877305-2019-06-02
- Chiu S. L., Chen C. M., Cline H. T. Insulin receptor signaling regulates synapse number, dendritic plasticity, and circuit function in vivo // Neuron. 2008. V. 58. №5. P. 708-719.
- DOI: 10.1016/j.neuron.2008.04.014
- Apostolatos A. et al. Insulin promotes neuronal survival via the alternatively spliced protein kinase CδII isoform // Journal of Biological Chemistry. 2012. V. 287. №12. P. 9299-9310.
- DOI: 10.1074/jbc.M111.313080
- Kleinridders A. et al. Insulin action in brain regulates systemic metabolism and brain function // Diabetes. 2014. V. 63. №7. P. 2232-2243.
- DOI: 10.2337/db14-0568
- Biessels G. J. et al. Dementia and cognitive decline in type 2 diabetes and prediabetic stages: towards targeted interventions // The lancet Diabetes & endocrinology. 2014. V. 2. №3. P. 246-255.
- DOI: 10.1016/S2213-8587(13)70088-3
- Vigneri R., Goldfine I. D., Frittitta L. Insulin, insulin receptors, and cancer // Journal of endocrinological investigation. 2016. V. 39. №12. P. 1365-1376.
- DOI: 10.1007/s40618-016-0508-7
- Derakhshan F., Toth C. Insulin and the brain // Current diabetes reviews. 2013. V. 9. №2. P. 102-116.
- DOI: 10.2174/157339913805076454
- Woods S. C. et al. Insulin and the blood-brain barrier //Current pharmaceutical design. 2003. V. 9. №10. P. 795.
- DOI: 10.2174/1381612033455323
- Martins J. P. et al. Communication from the periphery to the hypothalamus through the blood-brain barrier: an in vitro platform // International journal of pharmaceutics. 2016. V. 499. №1-2. P. 119-130.
- DOI: 10.1016/j.ijpharm.2015.12.058
- Devaskar S. U. et al. Insulin gene expression and insulin synthesis in mammalian neuronal cells // Journal of Biological Chemistry. 1994. V. 269. №11. P. 8445-8454.
- DOI: 10.1111/cns.12866
- Pomytkin I. et al. Insulin receptor in the brain: Mechanisms of activation and the role in the CNS pathology and treatment // CNS neuroscience & therapeutics. 2018. V. 24. №9. P. 763-774.
- DOI: 10.1111/cns.12866
- Belfiore A. et al. Insulin receptor isoforms in physiology and disease: an updated view // Endocrine reviews. 2017. V. 38. №5. P. 379-431.
- DOI: 10.1210/er.2017-00073
- Belfiore A. et al. Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease // Endocrine reviews. 2009. V. 30. №6. P. 586-623.
- DOI: 10.1210/er.2008-0047
- Hölscher C. New drug treatments show neuroprotective effects in Alzheimer's and Parkinson's diseases // Neural regeneration research. 2014. V. 9. №21. P. 1870.
- DOI: 10.4103/1673-5374.145342
- Akintola A. A., van Heemst D. Insulin, aging, and the brain: mechanisms and implications // Frontiers in endocrinology. 2015. V. 6. P. 13.
- DOI: 10.3389/fendo.2015.00013
- Numan S., Russell D. S. Discrete expression of insulin receptor substrate-4 mRNA in adult rat brain // Molecular brain research. 1999. V. 72. №1. P. 97-102.
- DOI: 10.1016/S0169-328X(99)00160-6
- Araki E. et al. Signalling in mice with targeted disruption // Nature. 1994. V. 372. №1. P. 186-90.
- Schubert M. et al. Insulin receptor substrate-2 deficiency impairs brain growth and promotes tau phosphorylation // Journal of Neuroscience. 2003. V. 23. №18. P. 7084-7092.
- DOI: 10.1523/JNEUROSCI.23-18-07084.2003
- Taguchi A., Wartschow L. M., White M. F. Brain IRS2 signaling coordinates life span and nutrient homeostasis // Science. 2007. V. 317. №5836. P. 369-372.
- DOI: 10.1126/science.1142179
- Sadagurski M. et al. Irs2 and Irs4 synergize in non-LepRb neurons to control energy balance and glucose homeostasis // Molecular metabolism. 2014. V. 3. №1. P. 55-63.
- DOI: 10.1016/j.molmet.2013.10.004
- Brüning J. C. et al. Role of brain insulin receptor in control of body weight and reproduction // Science. 2000. V. 289. №5487. P. 2122-2125.
- DOI: 10.1126/science.289.5487.2122
- Bomfim T. R. et al. An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer's disease-associated Aβ oligomers // The Journal of clinical investigation. 2012. V. 122. №4. P. 1339-1353.
- DOI: 10.1172/JCI57256
- Talbot K. et al. Demonstrated brain insulin resistance in Alzheimer's disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline // The Journal of clinical investigation. 2012. V. 122. №4. P. 1316-1338.
- DOI: 10.1172/JCI59903
- Denver P., English A., McClean P. L. Inflammation, insulin signaling and cognitive function in aged APP/PS1 mice // Brain, behavior, and immunity. 2018. V. 70. P. 423-434.
- DOI: 10.1016/j.bbi.2018.03.032
- Boucher J., Kleinridders A., Kahn C. R. Insulin receptor signaling in normal and insulin-resistant states // Cold Spring Harbor perspectives in biology. 2014. V. 6. №1. P. a009191.
- DOI: 10.1101/cshperspect.a009191
- Cho N. H. et al. IDF Diabetes Atlas: Global estimates of diabetes prevalence for 2017 and projections for 2045 // Diabetes research and clinical practice. 2018. V. 138. P. 271-281.
- DOI: 10.1016/j.diabres.2018.02.023
- Pugazhenthi S., Qin L., Reddy P. H. Common neurodegenerative pathways in obesity, diabetes, and Alzheimer's disease // Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease. 2017. V. 1863. №5. P. 1037-1045.
- DOI: 10.1016/j.bbadis.2016.04.017
- Reitz C., Mayeux R. Alzheimer disease: epidemiology, diagnostic criteria, risk factors and biomarkers // Biochemical pharmacology. 2014. V. 88. №4. P. 640-651.
- DOI: 10.1016/j.bcp.2013.12.024
- Anor C. J. et al. Neuropsychiatric symptoms in Alzheimer disease, vascular dementia, and mixed dementia // Neurodegenerative Diseases. 2017. V. 17. №4-5. P. 127-134.
- DOI: 10.1159/000455127
- McGeer P. L., McGeer E. G. The amyloid cascade-inflammatory hypothesis of Alzheimer disease: implications for therapy // Acta neuropathologica. 2013. V. 126. №4. P. 479-497.
- DOI: 10.1007/s00401-013-1177-7
- Wright A. L. et al. Neuroinflammation and neuronal loss precede Aβ plaque deposition in the hAPP-J20 mouse model of Alzheimer's disease // PloS one. 2013. V. 8. №4.
- DOI: 10.1371/journal.pone.0059586
- Li J. et al. Effects of diabetes mellitus on cognitive decline in patients with Alzheimer disease: a systematic review // Canadian journal of diabetes. 2017. V. 41. №1. P. 114-119.
- DOI: 10.1016/j.jcjd.2016.07.003
- Sripetchwandee J., Chattipakorn N., Chattipakorn S. C. Links between obesity-induced brain insulin resistance, brain mitochondrial dysfunction, and dementia // Frontiers in endocrinology. 2018. V. 9. P. 496.
- DOI: 10.3389/fendo.2018.00496
- Diaz A., Escobedo C., Treviño S., Chávez R., Lopez-Lopez G., Moran C.,.. Muñoz-Arenas G. Metabolic syndrome exacerbates the recognition memory impairment and oxidative-inflammatory response in rats with an intrahippocampal injection of amyloid beta // Oxidative medicine and cellular longevity. 2018. P. 1-42.
- DOI: 10.1155/2018/1358057
- Treviño S. et al. A high calorie diet causes memory loss, metabolic syndrome and oxidative stress into hippocampus and temporal cortex of rats // Synapse. 2015. V. 69. №9. P. 421-433.
- DOI: 10.1002/syn.21832
- Pierce A. L., Bullain S. S., Kawas C. H. Late-onset Alzheimer disease // Neurologic clinics. 2017. V. 35. №2. P. 283-293.
- DOI: 10.1016/j.ncl.2017.01.006
- Yin F. et al. Energy metabolism and inflammation in brain aging and Alzheimer's disease // Free Radical Biology and Medicine. 2016. V. 100. P. 108-122.
- DOI: 10.1016/j.freeradbiomed.2016.04.200
- Denver P., McClean P. L. Distinguishing normal brain aging from the development of Alzheimer's disease: inflammation, insulin signaling and cognition // Neural regeneration research. 2018. V. 13. №10. P. 1719.
- DOI: 10.4103/1673-5374.238608
- Frölich L. et al. Brain insulin and insulin receptors in aging and sporadic Alzheimer's disease // Journal of neural transmission. 1998. V. 105. №4-5. P. 423-438.
- DOI: 10.1007/s007020050068
- Ratzmann K. P., Hampel R. Glucose and insulin concentration patterns in cerebrospinal fluid following intravenous glucose injection in humans // Endokrinologie. 1980. V. 76. №2. P. 185-188. PMID:
- ISBN: 7004864
- Querfurth H. W., LaFerla F. M. Mechanisms of disease // N Engl J Med. 2010. V. 362. №4. P. 329-344.
- Suzanne M. Insulin resistance and neurodegeneration: progress towards the development of new therapeutics for Alzheimer's disease // Drugs. 2017. V. 77. №1. P. 47-65.
- DOI: 10.1007/s40265-016-0674-0
- Gabuzda D. et al. Inhibition of energy metabolism alters the processing of amyloid precursor protein and induces a potentially amyloidogenic derivative // Journal of Biological Chemistry. 1994. V. 269. №18. P. 13623-13628.
- Gasparini L. et al. Stimulation of β-amyloid precursor protein trafficking by insulin reduces intraneuronal β-amyloid and requires mitogen-activated protein kinase signaling // Journal of Neuroscience. 2001. V. 21. №8. P. 2561-2570.
- DOI: 10.1523/JNEUROSCI.21-08-02561.2001
- Nisbet R. M. et al. Tau aggregation and its interplay with amyloid-β // Acta neuropathologica. 2015. V. 129. №2. P. 207-220.
- DOI: 10.1007/s00401-014-1371-2
- Zimbone S. et al. Amyloid Beta monomers regulate cyclic adenosine monophosphate response element binding protein functions by activating type-1 insulin-like growth factor receptors in neuronal cells // Aging cell. 2018. V. 17. №1. P. e12684.
- DOI: 10.1111/acel.12684
- Ling X. et al. Amyloid beta antagonizes insulin promoted secretion of the amyloid beta protein precursor // Journal of Alzheimer's disease. 2002. V. 4. №5. P. 369-374.
- DOI: 10.3233/JAD-2002-4504
- Zhao W. Q. et al. Amyloid beta oligomers induce impairment of neuronal insulin receptors // The FASEB Journal. 2008. V. 22. №1. P. 246-260.
- DOI: 10.1096/fj.06-7703com
- Ma Q. L. et al. β-amyloid oligomers induce phosphorylation of tau and inactivation of insulin receptor substrate via c-Jun N-terminal kinase signaling: suppression by omega-3 fatty acids and curcumin // Journal of Neuroscience. 2009. V. 29. №28. P. 9078-9089.
- DOI: 10.1523/JNEUROSCI.1071-09.2009
- Schubert M. et al. Role for neuronal insulin resistance in neurodegenerative diseases // Proceedings of the National Academy of Sciences. 2004. V. 101. №9. P. 3100-3105.
- DOI: 10.1073/pnas.0308724101
- Vandal M. et al. Insulin reverses the high-fat diet-induced increase in brain Aβ and improves memory in an animal model of Alzheimer disease // Diabetes. 2014. V. 63. №12. P. 4291-4301.
- DOI: 10.2337/db14-0375
- Farris W. et al. Insulin-degrading enzyme regulates the levels of insulin, amyloid β-protein, and the β-amyloid precursor protein intracellular domain in vivo // Proceedings of the National Academy of Sciences. 2003. V. 100. №7. P. 4162-4167.
- DOI: 10.1073/pnas.0230450100
- Ittner L. M. et al. Dendritic function of tau mediates amyloid-β toxicity in Alzheimer's disease mouse models // Cell. 2010. V. 142. №3. P. 387-397.
- DOI: 10.1016/j.cell.2010.06.036
- Hong M., Lee V. M. Y. Insulin and insulin-like growth factor-1 regulate tau phosphorylation in cultured human neurons // Journal of Biological Chemistry. 1997. V. 272. №31. P. 19547-19553.
- DOI: 10.1074/jbc.272.40.25326
- Bhat R. et al. Structural insights and biological effects of glycogen synthase kinase 3-specific inhibitor AR-A014418 // Journal of Biological Chemistry. 2003. V. 278. №46. P. 45937-45945.
- DOI: 10.1074/jbc.M306268200
- Freude S. et al. Peripheral hyperinsulinemia promotes tau phosphorylation in vivo // Diabetes. 2005. V. 54. №12. P. 3343-3348.
- DOI: 10.2337/diabetes.54.12.3343
- Cheng C. M. et al. Tau is hyperphosphorylated in the insulin-like growth factor-I null brain // Endocrinology. 2005. V. 146. №12. P. 5086-5091.
- DOI: 10.1210/en.2005-0063
- Phiel C. J. et al. GSK-3α regulates production of Alzheimer's disease amyloid-β peptides // Nature. 2003. V. 423. №6938. P. 435-439.
- DOI: 10.1038/nature01640
- Sims-Robinson C. et al. How does diabetes accelerate Alzheimer disease pathology? // Nature Reviews Neurology. 2010. V. 6. №10. P. 551.
- DOI: 10.1038/nrneurol.2010.130
- De la Monte S. M. et al. Neuronal thread protein regulation and interaction with microtubule-associated proteins in SH-Sy5y neuronal cells // Cellular and Molecular Life Sciences CMLS. 2003. V. 60. №12. P. 2679-2691.
- DOI: 10.1007/s00018-003-3305-3
- Bedse G. et al. Aberrant insulin signaling in Alzheimer's disease: current knowledge // Frontiers in neuroscience. 2015. V. 9. P. 204.
- DOI: 10.3389/fnins.2015.00204
- Zlokovic B. V. Neurovascular pathways to neurodegeneration in Alzheimer's disease and other disorders // Nature Reviews Neuroscience. 2011. V. 12. №12. P. 723-738.
- DOI: 10.1038/nrn3114
- Kahn A. M. et al. Insulin Acutely Inhibits Cultured Vascular Smooth Muscle Cell Contraction by a Nitric Oxide Synthase-Dependent Pathway // Hypertension. 1997. V. 30. №4. P. 928-933.
- DOI: 10.1161/01.HYP.30.4.928
- Bhamra M. S., Ashton N. J. Finding a pathological diagnosis for A lzheimer's disease: Are inflammatory molecules the answer? // Electrophoresis. 2012. V. 33. №24. P. 3598-3607.
- DOI: 10.1002/elps.201200161
- Mushtaq G. et al. Alzheimer's disease and type 2 diabetes via chronic inflammatory mechanisms // Saudi journal of biological sciences. 2015. V. 22. №1. P. 4-13.
- DOI: 10.1016/j.sjbs.2014.05.003
- Fishel M. A. et al. Hyperinsulinemia provokes synchronous increases in central inflammation and β-amyloid in normal adults // Archives of neurology. 2005. V. 62. №10. P. 1539-1544.
- DOI: 10.1001/archneur.62.10.noc50112
- Sokolova A. et al. Monocyte chemoattractant protein-1 plays a dominant role in the chronic inflammation observed in Alzheimer's disease // Brain pathology. 2009. V. 19. №3. P. 392-398.
- DOI: 10.1111/j.1750-3639.2008.00188.x
- Nakamura M., Watanabe N. Ubiquitin-like protein MNSFβ/endophilin II complex regulates Dectin-1-mediated phagocytosis and inflammatory responses in macrophages // Biochemical and biophysical research communications. 2010. V. 401. №2. P. 257-261.
- DOI: 10.1016/j.bbrc.2010.09.045
- Akash M. S. H., Rehman K., Chen S. Role of inflammatory mechanisms in pathogenesis of type 2 diabetes mellitus // Journal of cellular biochemistry. 2013. V. 114. №3. P. 525-531.
- DOI: 10.1002/jcb.24402
- Erickson M. A., Hansen K., Banks W. A. Inflammation-induced dysfunction of the low-density lipoprotein receptor-related protein-1 at the blood-brain barrier: protection by the antioxidant N-acetylcysteine // Brain, behavior, and immunity. 2012. V. 26. №7. P. 1085-1094.
- DOI: 10.1016/j.bbi.2012.07.003
- Matrone C. et al. Inflammatory risk factors and pathologies promoting Alzheimer's disease progression: is RAGE the key // Histology and histopathology. 2015. V. 30. №2. P. 125-139.
- Münch G. et al. Alzheimer's disease-synergistic effects of glucose deficit, oxidative stress and advanced glycation endproducts // Journal of neural transmission. 1998. V. 105. №4-5. P. 439-461.
- DOI: 10.1007/s007020050069
- Deane R. et al. RAGE mediates amyloid-β peptide transport across the blood-brain barrier and accumulation in brain // Nature medicine. 2003. V. 9. №7. P. 907-913.
- DOI: 10.1038/nm890
- Chiang M. C. et al. Metformin activation of AMPK-dependent pathways is neuroprotective in human neural stem cells against Amyloid-beta-induced mitochondrial dysfunction // Experimental cell research. 2016. V. 347. №2. P. 322-331.
- DOI: 10.1016/j.yexcr.2016.08.013
- Chung M. M. et al. The neuroprotective role of metformin in advanced glycation end product treated human neural stem cells is AMPK-dependent // Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease. 2015. V. 1852. №5. P. 720-731.
- DOI: 10.1016/j.bbadis.2015.01.006
- Gupta A., Bisht B., Dey C. S. Peripheral insulin-sensitizer drug metformin ameliorates neuronal insulin resistance and Alzheimer's-like changes // Neuropharmacology. 2011. V. 60. №6. P. 910-920.
- DOI: 10.1016/j.neuropharm.2011.01.033
- Kickstein E. et al. Biguanide metformin acts on tau phosphorylation via mTOR/protein phosphatase 2A (PP2A) signaling // Proceedings of the National Academy of Sciences. 2010. V. 107. №50. P. 21830-21835.
- DOI: 10.1073/pnas.0912793107
- Li J. et al. Metformin attenuates Alzheimer's disease-like neuropathology in obese, leptin-resistant mice // Pharmacology biochemistry and behavior. 2012. V. 101. №4. P. 564-574.
- DOI: 10.1016/j.pbb.2012.03.002
- Chen Y. et al. Antidiabetic drug metformin (GlucophageR) increases biogenesis of Alzheimer's amyloid peptides via up-regulating BACE1 transcription // Proceedings of the National Academy of Sciences. 2009. V. 106. №10. P. 3907-3912.
- DOI: 10.1073/pnas.0807991106
- Ng T. P. et al. Long-term metformin usage and cognitive function among older adults with diabetes // Journal of Alzheimer's Disease. 2014. V. 41. №1. P. 61-68.
- DOI: 10.3233/JAD-131901
- Guo M. et al. Metformin may produce antidepressant effects through improvement of cognitive function among depressed patients with diabetes mellitus // Clinical and experimental pharmacology and physiology. 2014. V. 41. №9. P. 650-656.
- DOI: 10.1111/1440-1681.12265
- Herath P. M. et al. The effect of diabetes medication on cognitive function: evidence from the PATH through life study // BioMed research international. 2016. V. 2016.
- DOI: 10.1155/2016/7208429
- Moore E. M. et al. Increased risk of cognitive impairment in patients with diabetes is associated with metformin // Diabetes care. 2013. V. 36. №10. P. 2981-2987.
- DOI: 10.2337/dc13-0229
- Osborne C. et al. Glimepiride protects neurons against amyloid-β-induced synapse damage // Neuropharmacology. 2016. V. 101. P. 225-236.
- DOI: 10.1016/j.neuropharm.2015.09.030
- Alp H. et al. Protective effects of beta glucan and gliclazide on brain tissue and sciatic nerve of diabetic rats induced by streptozosin // Experimental diabetes research. 2012. V. 2012.
- DOI: 10.1155/2012/230342
- Esmaeili M. H., Bahari B., Salari A. A. ATP-sensitive potassium-channel inhibitor glibenclamide attenuates HPA axis hyperactivity, depression-and anxiety-related symptoms in a rat model of Alzheimer's disease // Brain research bulletin. 2018. V. 137. P. 265-276.
- DOI: 10.1016/j.brainresbull.2018.01.001
- Hsu C. C. et al. Incidence of dementia is increased in type 2 diabetes and reduced by the use of sulfonylureas and metformin // Journal of Alzheimer's Disease. 2011. V. 24. №3. P. 485-493.
- DOI: 10.3233/JAD-2011-101524
- Landreth G. Therapeutic use of agonists of the nuclear receptor PPARγ in Alzheimer's disease // Current Alzheimer Research. 2007. V. 4. №2. P. 159-164.
- DOI: 10.2174/156720507780362092
- Heneka M. T. et al. Acute treatment with the PPARγ agonist pioglitazone and ibuprofen reduces glial inflammation and Aβ1-42 levels in APPV717I transgenic mice // Brain. 2005. V. 128. №6. P. 1442-1453.
- DOI: 10.1093/brain/awh452
- Yu Y. et al. Insulin sensitizers improve learning and attenuate tau hyperphosphorylation and neuroinflammation in 3xTg-AD mice // Journal of neural transmission. 2015. V. 122. №4. P. 593-606.
- DOI: 10.1007/s00702-014-1294-z
- Fernandez-Martos C. M. et al. Combination treatment with leptin and pioglitazone in a mouse model of Alzheimer's disease // Alzheimer's & Dementia: Translational Research & Clinical Interventions. 2017. V. 3. №1. P. 92-106.
- DOI: 10.1016/j.trci.2016.11.002
- Sato T. et al. Efficacy of PPAR-γ agonist pioglitazone in mild Alzheimer disease // Neurobiology of aging. 2011. V. 32. №9. P. 1626-1633.
- DOI: 10.1016/j.neurobiolaging.2009.10.009
- Cheng H. et al. The peroxisome proliferators activated receptor-gamma agonists as therapeutics for the treatment of Alzheimer's disease and mild-to-moderate Alzheimer's disease: a meta-analysis // International Journal of Neuroscience. 2016. V. 126. №4. P. 299-307.
- DOI: 10.3109/00207454.2015.1015722
- Hölscher C. The role of GLP-1 in neuronal activity and neurodegeneration // Vitamins & Hormones. Academic Press, 2010. V. 84. P. 331-354.
- DOI: 10.1016/B978-0-12-381517-0.00013-8
- Hunter K., Hölscher C. Drugs developed to treat diabetes, liraglutide and lixisenatide, cross the blood brain barrier and enhance neurogenesis // BMC neuroscience. 2012. V. 13. №1. P. 33.
- DOI: 10.1186/1471-2202-13-33
- McClean P. L., Hölscher C. Liraglutide can reverse memory impairment, synaptic loss and reduce plaque load in aged APP/PS1 mice, a model of Alzheimer's disease // Neuropharmacology. 2014. V. 76. P. 57-67.
- DOI: 10.1016/j.neuropharm.2013.08.005
- Hansen H. H. et al. The GLP-1 receptor agonist liraglutide reduces pathology-specific tau phosphorylation and improves motor function in a transgenic hTauP301L mouse model of tauopathy // Brain research. 2016. V. 1634. P. 158-170.
- DOI: 10.1016/j.brainres.2015.12.052
- Gejl M. et al. In Alzheimer's disease, 6-month treatment with GLP-1 analog prevents decline of brain glucose metabolism: randomized, placebo-controlled, double-blind clinical trial // Frontiers in aging neuroscience. 2016. V. 8. P. 108.
- DOI: 10.3389/fnagi.2016.00108
- Kosaraju J. et al. Saxagliptin: a dipeptidyl peptidase-4 inhibitor ameliorates streptozotocin induced Alzheimer's disease // Neuropharmacology. 2013. V. 72. P. 291-300.
- DOI: 10.1016/j.neuropharm.2013.04.008
- Kosaraju J. et al. Vildagliptin: an anti-diabetes agent ameliorates cognitive deficits and pathology observed in streptozotocin-induced Alzheimer's disease // Journal of Pharmacy and Pharmacology. 2013. V. 65. №12. P. 1773-1784.
- DOI: 10.1111/jphp.12148
- Kornelius E. et al. DPP-4 inhibitor linagliptin attenuates Aβ-induced cytotoxicity through activation of AMPK in neuronal cells // CNS neuroscience & therapeutics. 2015. V. 21. №7. P. 549-557.
- DOI: 10.1111/cns.12404
- Rizzo M. R. et al. Dipeptidyl peptidase-4 inhibitors have protective effect on cognitive impairment in aged diabetic patients with mild cognitive impairment // Journals of Gerontology Series A: Biomedical Sciences and Medical Sciences. 2014. V. 69. №9. P. 1122-1131.
- DOI: 10.1093/gerona/glu032
- Kern W. et al. Improving influence of insulin on cognitive functions in humans // Neuroendocrinology. 2001. V. 74. №4. P. 270-280.
- DOI: 10.1159/000054694
- Freiherr J. et al. Intranasal insulin as a treatment for Alzheimer's disease: a review of basic research and clinical evidence // CNS drugs. 2013. V. 27. №7. P. 505-514.
- DOI: 10.1007/s40263-013-0076-8
- Plum L., Schubert M., Brüning J. C. The role of insulin receptor signaling in the brain // Trends in Endocrinology & Metabolism. 2005. V. 16. №2. P. 59-65.
- DOI: 10.1016/j.tem.2005.01.008
- Craft S. et al. Effects of regular and long-acting insulin on cognition and Alzheimer's disease biomarkers: a pilot clinical trial // Journal of Alzheimer's Disease. 2017. V. 57. №4. P. 1325-1334.
- DOI: 10.3233/JAD-161256
- Łabuzek K. et al. Quantification of metformin by the HPLC method in brain regions, cerebrospinal fluid and plasma of rats treated with lipopolysaccharide // Pharmacological Reports. 2010. V. 62. №5. P. 956-965.
- DOI: 10.1016/S1734-1140(10)70357-1
- Cheng C. et al. Type 2 diabetes and antidiabetic medications in relation to dementia diagnosis // Journals of Gerontology Series A: Biomedical Sciences and Medical Sciences. 2014. V. 69. №10. P. 1299-1305.
- DOI: 10.1093/gerona/glu073
- Luchsinger J. A. et al. Metformin in amnestic mild cognitive impairment: results of a pilot randomized placebo controlled clinical trial // Journal of Alzheimer's Disease. 2016. V. 51. №2. P. 501-514.
- DOI: 10.3233/JAD-150493
- Luchsinger J. A. et al. Metformin, lifestyle intervention, and cognition in the diabetes prevention program outcomes study // Diabetes care. 2017. V. 40. №7. P. 958-965.
- DOI: 10.2337/dc16-2376
- Valencia W. M. et al. Metformin and ageing: improving ageing outcomes beyond glycaemic control // Diabetologia. 2017. V. 60. №9. P. 1630-1638.
- DOI: 10.1007/s00125-017-4349-5
- Imfeld P. et al. Metformin, other antidiabetic drugs, and risk of Alzheimer's disease: a population-based case-control study // Journal of the American Geriatrics Society. 2012. V. 60. №5. P. 916-921.
- DOI: 10.1111/j.1532-5415.2012.03916.x
- Geldmacher D. S. et al. A randomized pilot clinical trial of the safety of pioglitazone in treatment of patients with Alzheimer disease // Archives of neurology. 2011. V. 68. №1. P. 45-50.
- DOI: 10.1001/archneurol.2010.229
- Femminella G. D. et al. Antidiabetic drugs in Alzheimer's disease: Mechanisms of action and future perspectives // Journal of diabetes research. 2017. V. 2017.
- DOI: 10.1155/2017/7420796
- Nauck M. A. Glucagon-like peptide 1 (GLP-1) in the treatment of diabetes // Hormone and metabolic research. 2004. V. 36. №11/12. P. 852-858.
- DOI: 10.1055/s-2004-826175
- Drucker D. J. et al. Incretin-based therapies for the treatment of type 2 diabetes: evaluation of the risks and benefits // Diabetes care. 2010. V. 33. №2. P. 428-433.
- DOI: 10.2337/dc09-1499
- Cork S. C. et al. Distribution and characterization of Glucagon-like peptide-1 receptor expressing cells in the mouse brain // Molecular metabolism. 2015. V. 4. №10. P. 718-731.
- DOI: 10.1016/j.molmet.2015.07.008
- Liu X. Y. et al. Liraglutide prevents beta-amyloid-induced neurotoxicity in SH-SY5Y cells via a PI3K-dependent signaling pathway // Neurological research. 2016. V. 38. №4. P. 313-319.
- DOI: 10.1080/01616412.2016.1145914
- Cai H. Y. et al. Lixisenatide attenuates the detrimental effects of amyloid β protein on spatial working memory and hippocampal neurons in rats // Behavioural brain research. 2017. V. 318. P. 28-35.
- DOI: 10.1016/j.bbr.2016.10.033
- Isik A. T. et al. The effects of sitagliptin, a DPP-4 inhibitor, on cognitive functions in elderly diabetic patients with or without Alzheimer's disease // Diabetes research and clinical practice. 2017. V. 123. P. 192-198.
- DOI: 10.1016/j.diabres.2016.12.010
- Shingo A. S. et al. Intracerebroventricular administration of an insulin analogue recovers STZ-induced cognitive decline in rats // Behavioural brain research. 2013. V. 241. P. 105-111.
- DOI: 10.1016/j.bbr.2012.12.005
