Исследование влияния концентрации лития на скорость диффузии в кристаллическом кремнии

Автор: Попов З.И., Федоров А.С., Кузубов А.А., Елисеева Н.С.
Журнал: Сибирский аэрокосмический журнал @vestnik-sibsau
Рубрика: Технологические процессы и материалы
Статья в выпуске: 2 (48), 2013 года.
Бесплатный доступ
Квантово-химическим методом DFT проведено исследование влияния концентрации лития на высоту потенциального барьера перескока атома лития в кристаллическом кремнии. С использованием этих данных предложен новый метод расчета коэффициента диффузии лития в кристаллическом кремнии при различной концентрации лития SiLi x, где x изменяется в диапазоне от 0,05 до 0,75.
Кремний, литий, первопринципные расчеты, диффузия
Короткий адрес: https://sciup.org/148177064
IDR: 148177064 | УДК: 544.18
Influence of lithium concentration on the diffusion rate in crystalline silicon
Influence of concentration of lithium on the height of the potential junction barrier of lithium atoms in crystalline silicon was investigated with quantum-chemical DFT method. Using this data, a new method for calculation of lithium diffusion in crystalline silicon was proposed, with different concentrations of lithium SiLi x where x ranges from 0,05 to 0,75.
Текст научной статьи Исследование влияния концентрации лития на скорость диффузии в кристаллическом кремнии
Развитие литий-ионных аккумуляторов в настоящее время является одной из самых актуальных задач в области источников тока ввиду их перспективности, широкого использования в электронных устройствах и даже в автомобилестроении ближайшего будущего. По сравнению с другими перезаряжаемыми батареями, такими как кислотно-свинцовые, никель кадмиевые и никель-металл-гидридные, литий-йонные батареи обладают большим удельным зарядом, рабочим напряжением и меньшим током саморазряда. На сегодняшний день основным материалом анодов современных батарей является графит, который обладает адсорбционной емкостью по литию 372 мА·ч/г, а также материалы на его основе. При внедрении ионы лития слегка раздвигают слои углеродной матрицы и располагаются между ними, образуя интеркала-ты. Ввиду достаточно большого расстояния между слоями sp2-гибридизированного углерода, обеспечиваемого слабым ван-дер-ваальсовым взаимодействием, удельный объем углеродных материалов в процессе интеркаляции-деинтеркаляции ионов лития меняется незначительно, что является положительной чертой данных материалов. К сожалению, в таких материалах происходит значительный перенос электрического заряда с атомов лития на слои графита, что ведет к существенному отталкиванию ионов лития. Это приводит к тому, что максимальное содержание лития в графите соответствует фазе LiC6, соответствующей вышеупомянутой адсорбционной емкости. Однако уже известно, что теоретическая емкость для батарей с кристаллическим кремниевым анодом может составить величину в 10 раз большую, чем для углеродных материалов [1], поэтому материалы на основе кремния сейчас широко изучаются как анодный материал для следующего поколения литий-йонных батарей [2; 3]. Одним из важных аспектов исследования кремния в качестве анода является исследование диффузии лития внутри его кристаллической структуры.
В отличие от случая диффузии, в кристаллических средах с достаточно низкими потенциальными барьерами ( V barrier ~ (1÷10)·кT) для прыжков частиц, когда можно применять метод молекулярной динамики (MD), в структурах с высокими потенциальными барьерами V barrier , например в ковалентно связанных кристаллических структурах ( V barrier >> кT), частота перескоков атомов через потенциальные барьеры является чрезвычайно малой (rare events problem). Поэтому такие системы не поддаются прямому исследованию методом MD. Для ускорения моделирования процессов активационных перескоков в таких случаях были разработаны методики смещающего потенциала (bias potential) [4], метод гипердинамики [5], а также метод температурно-ускоренной динамики (temperature-accelerated dynamics) [6]. К сожалению, применение данных методик для реальных систем сдерживается сложностью интегрирования данных методик в стандартные MD алгоритмы и сложностью определения потенциального профиля для движения диффунди-руемых частиц. Особенно это касается описания диффузии в аморфных веществах, когда имеется бесконечное многообразие потенциальных профилей и барьеров для диффузии частиц.
Целью данной работы являлась разработка метода расчета диффузии примеси на основе данных, полученных из квантово-химических расчетов, и применение данного метода для исследования влияния концентрации атомов лития на скорость его диффузии.
Квантово-химические расчеты в работе проводились в рамках метода функционала плотности (DFT) [7] с градиентными поправками Perdew-Burke-Ernzerhof (PBE) [8] с коррекцией Grimme, учитывающей ванн-дер-ваальсово взаимодействие [9] в лицензионном пакете VASP (Vienna Ab-initio Simulation Package) [10–13]. Для эффективного уменьшения количества базисных функций и увеличения скорости расчетов в программе для всех атомов использовались псевдопотенциалы Вандербильта (Vanderbilt ultrasoft pseudopotential) [14]. Для выбора оптимальных параметров расчета и точности интегрирования по первой зоне Бриллюэна был использован набор к-точек, сгенерированный с помощью метода Монхорста-Пака [15] на равномерной сетке 3X3X3, так как было обнаружено, что благодаря большому размеру суперячейки увеличение числа к-точек не приводит к изменению энергии связи и электронной структуры, но значительно увеличивает время расчетов. Для проведения оптимизации геометрии координаты всех атомов в суперячейках варьировались с помощью метода сопряженных градиентов, используя вычисление сил, действующих на атомы. При этом оптимизация проводилась до тех пор, пока силы, действующие на каждый атом, не становились менее 0,05 эВ/Å.
Для расчетов была использована суперячейка кремния в виде куба, состоящая из 216 атомов. Кремний имеет алмазоподобную кристаллическую структуру, в которой атомы лития могут располагаться в тетраэдрических порах данной структуры, максимальное число таких пор, в которых могут находиться атомы лития, равно числу атомов кремния. В данную ячейку помещали от одного до четырех атомов лития, причем их расположение выбиралось таким образом, чтобы расстояние между ними было минимальным, и проводили оптимизацию геометрии. После этого один из атомов лития помещали в соседнюю к нему пустую пору (в ячейках, где больше двух литиев, изменяли положение лития, равноудаленного от других) и оптимизировали полученную ячейку. Далее была рассчитана высота потенциального барьера (переходное состояние) для прыжков атома лития между двумя оптимизированными состояниями. Данные расчеты были выполнены методом эластичной упругой ленты (NEB) [16]. При этом были рассчитаны траектория, потенциальный профиль и величина барьера перехода атома лития через седловую точку потенциального профиля. Высота потенциальных барьеров перескока атома лития с различным заполнением окружения представлены в таблице.
Высота потенциального барьера перескока атома лития в зависимости от количества атомов лития в ячейке LixSi216
|
Количество атомов Li |
1 |
2 |
3 |
4 |
|
Высота барьера, эВ |
0,903 |
0,749 |
0,477 |
0,362 |
Полученные данные были использованы для получения полиноминальной зависимости высоты барьера перескока атома лития от степени заполнения соседних к нему пор такими же атомами:
V barrier = A E ь о - 0,07 P 3 + 0,277 P 2 - 0,375 P , (1)
где A E b 0 = 0,903 эВ (рассчитанное значение высоты барьера перескока атома лития из одной тетраэдрической поры в соседнюю без окружающих атомов лития).
Сумма вероятностей заполнения атомами лития пор окружающих ячейку, из которой совершается перескок, за исключением той, в которую совершается перескок ( m = 1,2,3), составляет
P = Z P m . (2)
Для вычисления коэффициента диффузии была предложена эффективная методика, основанная на вычислении временной эволюции распределения атомов лития P ( R i , t n ) в дискретном времени t n и пространстве R i . Данная эволюция описывается дискретным (заменяющим непрерывное уравнение диффузии) производящим уравнением (master equation) марковского процесса для степени заполнения узла i в момент времени t k :
D dP(Ri, tn ) _ X 1 Hi D D \Di D * МЛ Di D * \\
----”----= A W (Ri, Rj ) P(Rj , tn )(1 - P (Ri, tn )) - dt Rj (3)
D D D D
- W(Rj,Ri)P(Ri,tn)(1 -P(Rj,tn)), здесь W(Ri,Rj) – частота перескока атома примеси из минимума R j в минимум Ri . Данные величины для прыжка атома в любом направлении Ri ^ Rj через барьер Vbarrier(Ri, Rj) при температуре T рассчитывались с помощью уравнения аррениусовского типа:
W (R i , Rj ) = W 0 exp f- V barrier ( R i , Rj) 1 . (4)
I kT J
Предэкспоненциальный множитель (эффективная частота колебаний) W 0 вычислялся с помощью известной формулы Веньярда [17], также учитывающего энергию нулевых колебаний E 0 :
W 0 =
3 N - 3 f - h v i
П 1 - e kT kT i = 1 | ?
Й 3 N - 4 f h v i
П 1 - ekT i = 1
где v ‘ - частота колебаний N атомов системы при нахождении перемещаемого атома в седловой точке, а ν i – в точке минимума.
После 100 итераций расчет распределения вероятности нахождения атомов лития в каждом из узлов выполнялся расчет коэффициента диффузии. Для этого численно решалось основное уравнение диффузии:
dc дx ’
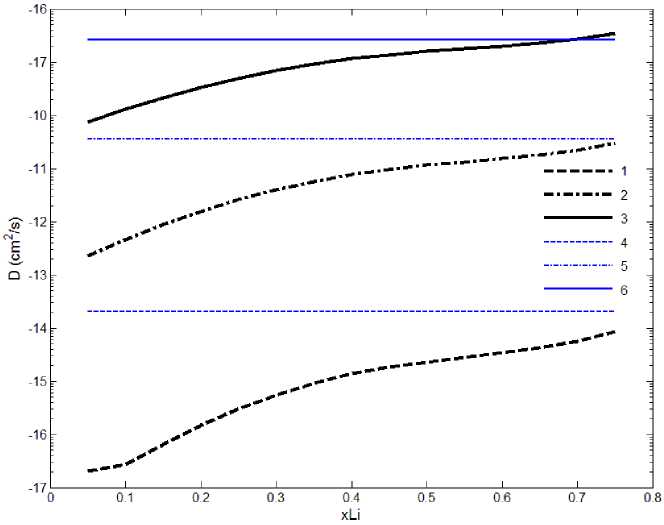
Зависимость коэффициента диффузии от концентрации лития в структуре LixSi:
при температуре: 1 – 273 К; 2 – 373 К; 3 – 473 К; экспериментальные данные: 4 – 273 К; 5 – 373 К; 6 – 473 К
здесь поток J – результирующее число атомов лития, прошедших в единицу времени через единичную площадь плоскости, перпендикулярной оси, вдоль которой направлен с – градиент концентрации атомов лития в этой плоскости (в числах атомов на единицу объема). Графики температурной зависимости коэффициентов диффузии с различной начальной концентрацией лития представлены на рисунке линиями с номерами 1, 2,3 для температуры 273, 373 и 473 К соответственно.
Линиями 4, 5, 6 показаны коэффициенты диффузии для аналогичных температур полученные из экспериментальной зависимости [18, 19]:
D = 2,5 ■ 10 - 3 ■ exp f2 0,^551211 ( k ■ T
В отличие от работы [20], в которой рассматривалась зависимость коэффициента диффузии от температуры, представленный алгоритм расчета позволил определить зависимость коэффициента диффузии как от температуры, так и от концентрации лития.
Итак, в ходе выполнения работы с помощью проведения первопринципных расчетов была получена зависимость коэффициента диффузии от концентрации лития и температуры в кристаллическом кремнии. Обнаружено значительное (на 1–2 порядка) увеличение скорости диффузии лития в кремнии при увеличении концентрации лития до состава Li 0,75 Si.
Referens
-
1. Kasavajjula U., Wang C. S., Appleby A. J. Nano-and bulk-silicon-based insertion anodes for lithium-ion secondary cells. J. Power Sources. 2007, vol. 163, 1003 p.
-
2. Winter M., Besenhard J. O. Electrochemical lithia-tion of tin and tin-based intermetallics and composites. Electrochim. Acta, 1999, vol. 45, 31 p.
-
3. Huggins R. A., Lithium alloy negative electrodes. J. Power Sources . 1999, vol. 81–82, 13 p.
-
4. Hamelberg D., Mongan J., McCammon J. A. Accelerated molecular dynamics: A promising and efficient simulation method for biomolecules. J. Chem. Phys . 2004, vol. 120, 11919 p.
-
5. Voter A. F. Hyperdynamics: Accelerated Molecular Dynamics of Infrequent Events. Phys. Rev. Lett. 1997, vol. 78, 3908 p.
-
6. Sorensen M. R., Voter A. F. Temperature-accelerated dynamics for simulation of infrequent event. J. Chem. Phys . 2000, vol. 112, 9599 p.
-
7. Kohn W., Sham L. J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, vol. 140, 1133 p.
-
8. Perdew J. P., Burke K., Ernzerhof M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, vol. 77, pp. 3865–3868.
-
9. Grimme S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comp. Chem. 2006, vol. 27, 1787 p.
-
10. Kresse G., Hafner J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B. 1993, vol. 47, № 1, pp. 558–561.
-
11. Kresse G., Hafner J. Ab initio molecular-dynamics simulation of the liquid-metal-amorphous-semiconductor transition in germanium. Phys. Rev. B. 1994, vol. 49, № 20, pp. 14251– 4269.
-
12. Kresse G., Furthmüller J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Computer Material Science. 1996, № 6, 15 p.
-
13. Kresse G., Furthmüller J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B. 1996, vol. 54, 11169 p.
-
14. Vanderbilt D. Soft self-consistent pseudopotentials in generalized eigenvalue formalism. Phys.Rev. B. 1990, vol. 41, 7892 p.
-
15. Monkhorst H. J., Pack J. D. Special points for Brillouin-zone integrations. Phys. Rev. B. 1976, vol. 13, pp. 5188–5192.
-
16. Henkelman G., Jonsson H. Improved tangent estimate in the nudged elastic band method for finding
minimum energy paths and saddle points. J. Chem. Phys . 2000, vol. 113, pp. 9978–9985.
-
17. Vineyard G. V. Frequency factors and isotope effects in solid state rate processes. J. Phys. Chem. Solids. 1957, vol. 3, pp. 121–127.
-
18. Pell E. M. Diffusion of Li in Si at High T and the Isotope Effect. Phys. Rev . 1960, vol. 119, № 3, pp. 1014–1021.
-
19. Pell E. M. Diffusion Rate of Li in Si at Low Temperature. Phys. Rev. 1960, vol. 119, № 4, pp. 1222–1225.
-
20. Fedorov, A. S., Popov Z. I., Kuzubov A. A., Ovchinnikov S. G. Teoreticheskiye issledovaniye diffuzii litiya v kristallicheskom i amorfnom kremnii (Theoretical study of lithium diffusion in crystalline and amorphous silicon). Pis'ma v ZHETF . 2012, vol. 95, pp. 159–163.
