Эндоэдральные комплексы углеродных нанотрубок с литием при конечных температурах

Автор: Созыкин Сергей Анатольевич, Бескачко Валерий Петрович
Рубрика: Физика
Статья в выпуске: 2 т.6, 2014 года.
Бесплатный доступ
Методами первопринципной молекулярной динамики моделируются строение и динамические свойства углеродных нанотрубок, содержащих в полости атомы лития, в диапазоне температур 100-700 К. Показано, что основные закономерности в строении таких комплексов, выявленные ранее при T = 0 K, сохраняются также и при всех рассмотренных температурах. Изучен характер движения внедренных атомов в зависимости от их концентрации и температуры. Предсказывается, что при малых концентрациях движение атомов лития становится инфинитным при температурах, превышающих комнатные. В отличие от этого случая, при концентрациях, превышающих порог кластерообразования, движение кластеров лития инфинитно даже при самых низких из рассмотренных температур.
Углеродные нанотрубки, эндоэдральный комплекс, атомная структура и динамика, компьютерное моделирование, молекулярная динамика, расчеты из первых принципов
Короткий адрес: https://sciup.org/147158815
IDR: 147158815 | УДК: 245.26
Endohedral complexes of carbon nanotubes with lithium at finite temperature
First principle molecular dynamics method is used to simulate the structure and dynamic properties of carbon nanotubes with lithium atoms in a cavity at temperature range of 100-700 K. It is shown that basic patterns in the structure of these complexes, revealed at T = 0 K, are kept at all considered temperatures. The character of the motion of interstitial atoms depending on their concentration and temperature is studied. It is predicted that movement of atoms at low lithium concentrations becomes infinite at temperatures greater than 300 K. On the contrary, at concentrations above the threshold of clustering, the movement of clusters of lithium is infinite at the lowest of considered temperatures.
Текст научной статьи Эндоэдральные комплексы углеродных нанотрубок с литием при конечных температурах
Эндоэдральным комплексом углеродной нанотрубки (УНТ) называется УНТ, содержащая в своей полости инородные атомные структуры. Такие комплексы принято обозначать как X@(n,m) , где X – символ, указывающий на внедренное вещество, а n и m – индексы хиральности трубки. С теоретической точки зрения такие объекты интересны тем, что имеют микроскопические размеры в двух измерениях и мезо- или даже макроскопические – в оставшемся измерении. Присутствие микроскопических измерений означает, что строение и свойства объекта должны описываться квантовомеханически, и поэтому результат взаимодействия УНТ с окружением нельзя предсказать так же просто, как для макроскопических тел. На данном этапе исследований, когда информации о свойствах комплексов еще недостаточно для ее полуэмпирического или феноменологического обобщения, целесообразным является использование априорных методов расчета, таких как метод функционала электронной плотности или ab initio молекулярной динамики.
Одними из самых простых и изученных теоретически являются комплексы с литием, Li@(n,m) , по причине, видимо, их перспективности для разработки нового поколения электрических батарей. В работе [1] рассмотрен комплекс Li@ (12,0), а в [2] – Li@ (5,5). Нами было изучено строение, электрические и механические свойства комплексов Li@ (7,7) и Li@ (11,0) [3–5]. Все эти исследования относятся к случаю низких температур, T ~ 0 K , в то время как реальные устройства обычно работают при температурах T ~ 300 K и выше. В настоящее время неясно, сохранятся ли выводы, сделанные в работах [1–5], при конечных, интересных для практики температурах. В настоящей работе делается попытка ответить на этот вопрос с помощью компьютерного моделирования обсуждаемых комплексов, исходя из первых физических принципов.
Модели комплексов
Во всех расчетах использовалась молекулярная динамика Кара–Паринелло, реализованная в пакете SIESTA [6]. Для обменно-корреляционного взаимодействия в приближении локальной плотности применялась параметризация Каперлея и Алдера. В модели 2 s 22 p 2 электроны углерода и 2 s 1 электрон лития рассматривались точно, а остальные описывались псевдопотенциалом Тро-улиера–Мартинса. Для валентных электронов использовался базис DZP (для C: две радиальные функции для 2 s и 2 p , и одна 3 d орбиталь; для Li: две радиальные функции для 2 s и одна 2 p орбиталь). Количество базисных функций, таким образом, составляло 13 для атома углерода и 3 для атома лития. В расчетах использовался термостат Нозе–Хувера.
Использованный нами метод, как и все априорные методы, является весьма ресурсоемким в
вычислительном плане, что существенно ограничивает как размер моделируемой системы, так и длину описываемой ею фазовой траектории. Так, при тех ресурсах, что предоставляются вычислительным кластером ЮУрГУ «Торнадо», возможно рассмотрение систем, состоящих из ~100 атомов углерода. Поэтому в качестве объекта исследования был выбран фрагмент комплекса Li@ (7,7) длиной ≈ 1 нм, содержащий 112 атомов углерода и от 1 до 4 атомов лития (рис. 1). Эта
модель изучалась в диапазоне температур 100–700 К. Использовались периодические граничные условия. Размер расчетной ячейки вдоль оси нанотрубки устанавливался в 10 раз меньше, чем размеры в двух оставшихся измерениях. Это позволяет считать, что моделируются макроскопически длинные комплексы, не взаимодействующие друг с другом.
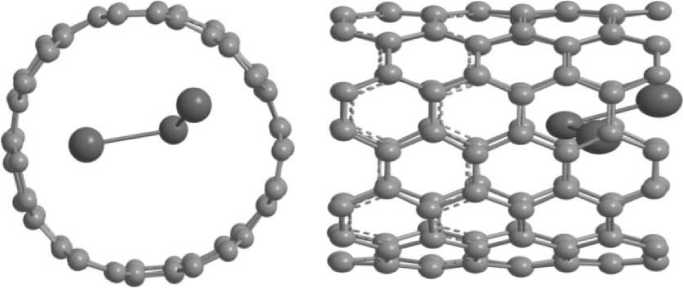
Рис.1. Модель фрагмента УНТ (7,7) c 3 атомами лития
Методика численных экспериментов
Указанный набор методов неоднократно успешно использовался нами [3–5] и другими исследователями при моделировании УНТ. Получаемые результаты, хотя и согласуются друг с другом, однако часто не вполне совпадают количественно. Ввиду сложности и громоздкости расчетов установить причины расхождений не просто. Всегда хочется иметь оценку потенциальной точности метода, той точности, которой можно достичь в расчетах более простых систем. С этой целью мы в настоящей работе рассмотрели две тестовые задачи о строении простых молекулярных систем, содержащих атомы Li и C: о молекуле бензола, взаимодействующей с атомом Li, LiC 6 H 6 , и о молекуле карбида лития, Li-C.
Несмотря на кажущуюся простоту, вопрос о равновесной структуре комплекса бензола с литием окончательно не решен [7]. Даже с помощью очень точных методов пока не удается выяснить, эквивалентны ли в нем длины связей С-С и Li-C. Ясно лишь, что атом Li находится практически над центром шестиугольника из атомов углерода, а расстояния Li-C находятся в диапазоне 0,215–0,265 нм [7]. Расчеты, выполненные нами с помощью описанных выше инструментов, показывают наличие трех различных пар расстояний Li-C: 0,224, 0,234 и 0,239 нм. Связи С-С распадаются на две группы: 4 связи имеют длину 0,143 нм и 2 связи – 0,138 нм. Для карбида лития мы рассчитали изменение полной энергии системы как функцию расстояния между атомами. Результаты представлены на рис. 2Рис. 2. Энергия карбида лития и они демонстрируют хорошее согласие с данными работы [8], полученными как с учетом всех электронов, так и с использованием псевдопотенциала. Таким образом, можно заключить, что использованная нами методика расчета больших молекулярных систем (с числом атомов порядка сотни) не уступает существенно по точности более совершенным методам, пригодным для описания малых систем.
Параметры процесса моделирования, ответственные за статистическую надежность результатов, подбирались в серии предварительных численных экспериментов. В качестве стартовых выбирались конфигурации, которым при температуре T = 0 соответствовал минимум энергии. Шаг по времени был взят равным 1 фс. При таком шаге уже через 100 шагов интегрирования уравнений движения диапазон флуктуаций температуры переставал изменяться, что позволяло считать температуру установившейся. Рис. 3 дает представление о характере флуктуаций температуры в установившемся режиме для комплекса УНТ (7,7) с 3 атомами лития.
1,5
данная работа полноэлектронный расчет, LDA [8]
псевдопотенциал (пакет ABINIT) [8]
СО
Ф
О
1,0
0,5
0,0
0,15 0,20
0,25 0,30
0,35
Длина связи C-Li, нм
Рис. 2. Энергия карбида лития как функция расстояния между атомами углерода и лития
Созыкин С.А., Бескачко В.П.
Эндоэдральные комплексы углеродных нанотрубок с литием при конечных температурах
Для оценки ряда свойств, например, электрического сопротивления, необходимо располагать ансамблем статистически независимых состояний системы, отвечающих данной температуре. Для определения характерного времени, спустя которое частицы «забывают» свое начальное состояние, вычислялась автокорреляционная функция скорости, ACF ( t )
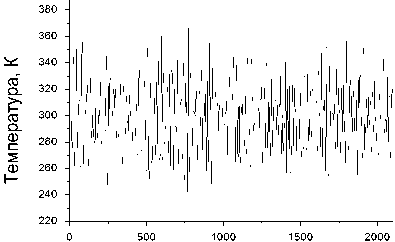
Количество шагов моделирования
Рис. 3. Флуктуации температуры во фрагменте комплекса Li@(7,7)
ACF(t)=〈Vi(0)Vi(t)〉 〈Vi(0)〉 , где угловые скобки означают усреднение по ансамблю частиц системы. Оказалось, что функция ACF(t) для атомов углерода убывает до нуля за 12 шагов моделирования (см. подробности ниже). Поэтому, если фазовая траектория системы построена за 1500 шагов моделирования (при этом время расчетов с помощью выбранных нами средств еще останется разумным), то это позволит создать ансамбль из 120–130 независимых состояний системы. Мы предполагаем, что этого будет достаточно, чтобы дать надежную оценку структуры и свойств комплексов. Заметим, что «разумное время вычислений» у нас составляет ~5 суток и достигается при использовании 120 вычислительных ядер Intel Xeon X5680 (3,33 ГГц).
Результаты
Из расчетов следует, что выявленные в [5] тенденции в строении комплексов при Т = 0 К сохраняются для всех рассмотренных температур: атомы Li преимущественно располагаются вблизи стенки УНТ, связываясь в кластеры, когда их число n в моделируемом фрагменте комплекса станет
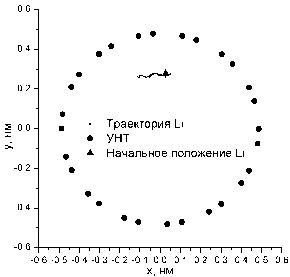
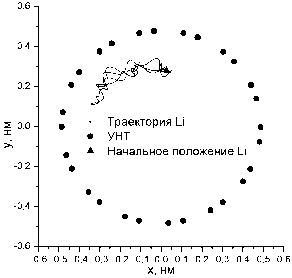
Рис. 4. Траектория атома лития (100 K – слева и 700 K – справа)
больше двух. Представление о характере движения внедренного атома дает рис. 4, где представ- лены проекции его траектории на плоскость, перпендикулярную оси нанотрубки. Из рис. 4 вид- но, что при малых температурах движение атома лития является финитным.
Время, фс Время, фс
1 атом 3 атома
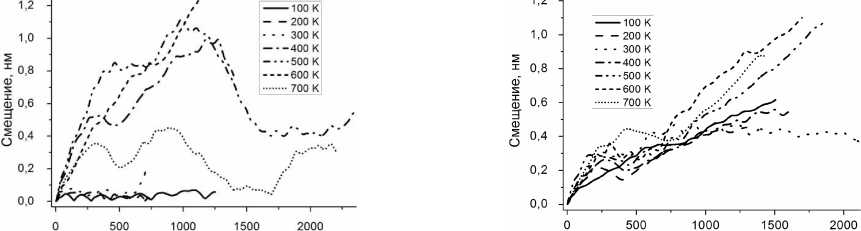
Рис. 5. Смещение атомов углерода и лития как функция времени при различных температурах
При больших температурах атом может удаляться из начального положения достаточно далеко, но и в этом случае расстояние от него до стенки нанотрубки изменяется мало (рис. 4). Более подробное исследование показывает (рис. 5), что при n = 1 движение атома остается финитным (атом локализован в окрестности определенного шестиугольника из атомов углерода) при температурах вплоть до 300 К и происходит в окрестности его равновесного при T = 0 положения. При более высоких температурах атом получает возможность переходить от одного шестиугольника к другому.
При n = 2 доступная для движения область несколько увеличивается, но при T < 300 K оста- ется ограниченной. Интересно отметить, что при n = 3, 4 движение образующихся кластеров не локализуется вблизи каких-либо мест углеродного каркаса УНТ даже при самой низкой из рас- смотренных температур (см. рис. 5).
Динамические характеристики внедренных атомов оценивались также с помощью ACF ( t ) (1). Стартовые конфигурации получались случайным помещением атома лития в цилиндрический слой на расстояниях от 0,25 до 0,31 нм от оси нанотрубки. На рис. 6 приведены результаты расчета таких функций для случаев, когда n изменяется от 1 до 4. Видно, что при n = 1 АФС в установившемся режиме движения испытывает незатухающие осцилляции с периодом около 100 фс, указывающие на колебательный характер движения атома Li около положений равновесия. С увеличением n амплитуда осцилляций уменьшается, так что при n = 4 остаются лишь случай-
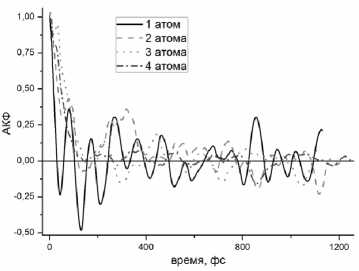
Рис. 6. Автокорреляционные функции скоростей атомов лития при 100 К
ные флуктуации АКФ, определяемые, скорее всего, статистикой при усреднении в (1). Это указывает на постепенное исчезновение колебательного режима движения и развитие движения вдоль поверхности трубки. Отметим, что и в этом случае движение носит сложных характер и не сводится к направленному движению в определенном направлении.
Заключение
Проведенные расчеты показывают, что
-
1) Выявленные в [5] при T = 0 тенденции структурообразования лития в комплексах Li@УНТ (преимущественное расположение невзаимодействующих между собой атомов Li вблизи стенки УНТ; образование кластеров Li при концентрациях более 2 ат. %) сохраняются и при конечных (до 700 К) температурах.
-
2) При низких температурах (до 300 К) и малых концентрациях (~1–2 ат. %) движение атомов лития локализуется в окрестности положений равновесия с редкими перескоками от одного положения к другому. При более высоких температурах движение становится инфинитным.
-
3) При концентрациях лития, превышающих порог кластерообразования ( C Li = 3 - 4), движение кластеров оказывается инфинитным даже при самых низких из рассмотренных температур из-за ослабления связи атомов кластера со стенкой УНТ.
Список литературы Эндоэдральные комплексы углеродных нанотрубок с литием при конечных температурах
- Theoretical study of adsorption of lithium atom on carbon nanotube/M. Senami, Y. Ikeda, A. Fukushima, A. Tachibana//AIP Advances. -2011. -Vol. 1. -P. 042106.
- Interaction and concerted diffusion of lithium in a (5,5) carbon nanotube/M. Khantha, N.A. Cordero, J.A. Alonso et al.//Physical review B. -2008. -Vol. 78. -P. 115430.
- Созыкин, С.А. Электрические свойства комплексов углеродной нанотрубки (7,7) с одиночными атомами Li, Na, S и Se/С.А. Созыкин, В.П. Бескачко//Вестник ЮУрГУ. Серия «Математика. Механика. Физика». -2012. -Вып. 7. -№ 34(293). -С. 113-119.
- Созыкин, С.А. Механические свойства комплексов углеродной нанотрубки (7,7) с одиночными атомами Li, Na, S и Se/С.А. Созыкин, В.П. Бескачко//Вестник ЮУрГУ. Серия «Математика. Механика. Физика». -2012. -Вып. 7. -№ 34(293). -С. 182-185.
- Sozykin, S.A. Structure of endohedral complexes of carbon nanotubes encapsulated with lithium and sodium/S.A. Sozykin, V.P. Beskachko//Molecular Physics. -2013. -Vol. 111. -Issue 7. -P. 930-938 DOI: 10.1080/00268976.2012.760049
- The Siesta method for ab initio order-N materials simulation/José M. Soler, Emilio Artacho, Julian D. Gale et al.//J. Phys.: Condens. Matter. -2002. -Vol. 14. -P. 2745-2779.
- Denis, P.A. C2V or C6V: Which is the most stable structure of the benzene-lithium complex?/P.A. Denis, F. Iribarne//Chemical Physics Letters. -2013. -Vol. 573. -P. 15-18.
- Electrical Transport in Single-Walled Carbon Nanotubes/M.J. Biercuk, S. Ilani, C.M. Marcus, and P.L. McEuen//Carbon Nanotubes, Topics in Applied Physics: сб. науч. тр. -2008. -Vol. 111. -P. 455-493.
