Клинический случай: новый вариант в гене NF1 у пациента с нейрофиброматозом 1-го типа

Автор: Жалсанова И.Ж., Фонова Е.А., Ербурова Д.Н., Петрова В.В., Киреева Т.Н., Сеитова Г.Н., Зернов Н.В., Скрябин Н.А.
Журнал: Сибирский онкологический журнал @siboncoj
Рубрика: Случай из клинической практики
Статья в выпуске: 3 т.25, 2026 года.
Бесплатный доступ
Актуальность. Нейрофиброматоз 1 типа распространенное наследственное заболевание с аутосомно-доминантным типом наследования, обусловленное патогенными генетическими вариантами в гене NF1, расположенном на хромосоме 17q11.2. Заболевание характеризуется высокой аллельной гетерогенностью и отсутствием четких гено-фенотипических корреляций, за исключением крупных делеций, ассоциированных с более тяжелым фенотипом. Клинически нейрофиброматоз характеризуется наличием множественных (>6) пятен цвета «кофе с молоком», нейрофибромами любого типа или плексиформными нейрофибромами, веснушками в подмышечных или паховых областях, гамартоматозными узелками Лиша радужной оболочки, глиомой зрительного нерва и костной дисплазией. В связи с этим описание каждого нового генетического варианта имеет существенное значение для расширения спектра известных вариантов и совершенствования молекулярной диагностики. Описание клинического случая. Описывается клиническое и молекулярно-генетическое обследование мальчика 10 лет, у которого с раннего детства отмечаются множественные пятна цвета «кофе с молоком», нарушение речи, гипотрофия I степени, рахит, последствия перинатального поражения ЦНС, миотонический синдром и темповая задержка моторного развития. При магнитно-резонансной томографии головного мозга выявлены признаки очагового поражения обоих полушарий, червя мозжечка, подкорковых ядер слева, выявлена глиома правого зрительного нерва. На основании клинических критериев диагностирован нейрофиброматоз 1 типа. Проведено полногеномное секвенирование ДНК с последующим биоинформатическим анализом и подтверждением результатов прямым секвенированием по Сэнгеру. Выявлен ранее не описанный сложный вероятно патогенный вариант NM_001042492.3:c.4060_4068 delinsC гена NF1 в гетерозиготном состоянии. Данный вариант приводит к сдвигу рамки считывания и преждевременной терминации трансляции после синтеза 23 аминокислот (p.Ser1354Leufs*23). Данный вариант отсутствует в доступных популяционных базах данных генетических вариантов. Заключение. В результате полногеномного секвенирования выявлен новый патогенный вариант в гене NF1, ассоциированный с развитием нейрофиброматоза 1 типа. Полученные данные расширяют спектр вариантов при данной патологии и могут быть использованы при медико-генетическом консультировании, а также в алгоритмах молекулярно-генетической диагностики пациентов с подозрением на нейрофиброматоз 1 типа.
Нейрофиброматоз, нейрофибромы, NF1, массовое параллельное секвенирование, пятна цвета «кофе с молоком», секвенирование генома, глиома зрительного нерва
Короткий адрес: https://sciup.org/140315699
IDR: 140315699 | УДК: 616-006.38.03:575.113.1 | DOI: 10.21294/1814-4861-2026-25-3-152-158
A novel variant of NF1 gene in a patient with neurofibromatosis type 1: a case report
Background. neurofibromatosis type 1 is a common hereditary autosomal dominant disorder caused by pathogenic genetic variants in the NF1 gene located on chromosome 17q11.2. the disease is characterized by high allelic heterogeneity and the absence of clear genotypic correlations, with the exception of large deletions associated with a more severe phenotype. Clinically, neurofibromatosis is characterized by the presence of multiple (>6) "café-au-lait” spots, neurofibromas of any type or plexiform neurofibromas, freckles in the axillary or inguinal areas, hamartomatous Lisch nodules of the iris, optic glioma, and bone dysplasia. therefore, the description of each new genetic variant is essential for expanding the spectrum of known variants and improving molecular diagnostics. Case Report. the article describes a clinical and molecular genetic study of a 10-year-old boy. since early childhood, the patient has had multiple “café-au-lait” spots, speech impairment, grade 1 hypotrophy, rickets, sequelae of perinatal CNs damage, myotonic syndrome, and a delay in motor development. Magnetic resonance imaging of the brain revealed signs of focal damage to both hemispheres, the cerebellar vermis, and the left subcortical nuclei, and a glioma of the right optic nerve was detected. Neurofibromatosis type 1 was diagnosed based on clinical criteria. Whole-genome DNA sequencing was performed, followed by bioinformatics analysis and confirmation of the results by direct sanger sequencing. A previously undescribed complex, likely pathogenic variant NM_001042492.3:c.40 60_4068delinsC of the NF1 gene was identified in the heterozygous state. this variant leads to a reading frameshift and premature translation termination after the synthesis of 23 amino acids (p.ser1354Leufs*23). this variant is not present in available population genetic variant databases. Conclusion. thus, wholegenome sequencing identified a new pathogenic variant in the NF1 gene associated with the development of neurofibromatosis type 1. the obtained data expand the range of variants for this pathology and can be used in medical genetic counseling, as well as in algorithms for molecular genetic diagnostics of patients with suspected neurofibromatosis type 1.
Текст научной статьи Клинический случай: новый вариант в гене NF1 у пациента с нейрофиброматозом 1-го типа
Нейрофиброматоз 1 типа (НФ1) является одним из наиболее распространенных факоматозов с аутосомно-доминантным типом наследования и высокой пенетрантностью, характеризующихся предрасположенностью к развитию доброкачественных и злокачественных опухолей нервной системы, а также множественными кожными, неврологическими и скелетными проявлениями [1]. Заболеваемость составляет приблизительно 1 случай на 2 500–3 000 новорожденных, причем около 50 % случаев обусловлены de novo мутациями [2, 3]. Данных по заболеваемости НФ1 в России нет ввиду отсутствия широкомасштабных эпидемиологических исследований. Учитывая уровень рождаемости, ежегодно в стране появляется на свет приблизительно 530 детей с данной патологией [4]. Нейрофиброматоз 1 типа характеризуется вы- раженным клиническим полиморфизмом, прогрессирующим течением, полиорганностью поражений и высокой частотой осложнений. В этой работе мы рассказываем о клиническом наблюдении пациента с симптомами НФ1. Для установления молекулярно-генетического диагноза заболевания пациенту и его матери проведено полногеномное секвенирование, в результате которого идентифицировали новый вероятно патогенный вариант NM_001042492.3:c.4060_4068delinsC гена NF1.
Материал и методы
В исследование включена семья с подозрением на нейрофиброматоз 1 типа у пробанда. Исследование проводилось с соблюдением этических норм в соответствии с Хельсинкской декларацией Всемирной медицинской ассоциации. Получено информированное согласие всех членов семьи на участие в обследовании. Исследование выполнено с использованием оборудования центра коллективного пользования «Медицинская геномика» на базе НИИ медицинской генетики Томского НИМЦ и одобрено комитетом по биомедицинской этике НИИ медицинской генетики Томского НИМЦ.
Массовое параллельное секвенирование образцов семьи проведено в рамках совместной научноисследовательской программы Национальная генетическая инициатива «100 000 + Я» на базе ООО «Биотехнологический кампус» с использованием технологии DNBSEQ.
Обработка данных секвенирования проведена согласно best practices GATK для «Germline SNPs Indels» с использованием набора программ GATK4. Оценка качества прочтений осуществлялась с помощью программы Qualimap. Выравнивание прочтений на целевые последовательности генов (GRCh38/hg38) осуществлялось с помощью BWA. Длина прочтений составила 2 × 101 п.н. Для каждого образца число прочтений Q10 составило не менее 90 % от числа прочтений, полученных в результате секвенирования. Для аннотирования использовалась программа Annovar. Для оценки клинической релевантности выявленных вариантов использованы базы данных OMIM, ClinVar, gnomAD v4.1.0 exomes, HPO и литературные данные. Секвенирование по Сэнгеру было проведено в обоих направлениях в области идентифицированного варианта на образцах ДНК пробанда и его матери. Были использованы следующие праймеры для полимеразной цепной реакции (ПЦР): прямой (5’-TTCCAATGAAGTCTACACGTTGC-3’); обратный (5’-ATTTCAGCCACATCCATCATACC-3’).
Описание клинического случая
В генетическую клинику НИИ медицинской генетики Томского НИМЦ обратились мать и сын с подозрением на нейрофиброматоз 1 типа. Мальчик, 10 лет, с жалобами на множественные пятна цвета «кофе с молоком» различной степени окрашивания и размера. У ребенка отмечаются нарушение речи, дефицит массы тела. Ребенок от третьей беременности, протекавшей на фоне угрозы прерывания в сроке 9 нед, эрозии шейки матки, острого респираторного заболевания средней степени тяжести, а также носительства вируса простого герпеса и цитомегаловируса. Роды вторые, срочные, на 39-й нед гестации. Масса тела при рождении составила 3320 г, длина – 51 см. Оценка по шкале Апгар – 8/8 баллов. В период новорожденности отмечалась желтушность кожных покровов, сохранявшаяся до 3 нед жизни. В возрасте 1 мес выявлены множественные гиперпигментированные пятна цвета «кофе с молоком» (café-au-lait) с последующей тенденцией к увеличению их количества. В возрасте двух лет пациент находился в отделении психоневрологии с основным клиническим диагнозом: нейрофибро- матоз 1 типа; сопутствующие: гипотрофия I степени, рахит, последствия перинатального поражения ЦНС, миотонический синдром и темповая задержка моторного развития. По результатам МРТ головного мозга выявлены признаки очагового поражения обоих полушарий, червя мозжечка, подкорковых ядер слева. Впервые описаны МР-признаки глиомы правого зрительного нерва. В динамике до 2022 г. при ежегодном мониторинге отрицательной динамики не отмечалось. По данным МРТ от 2023 г. отмечены увеличение размеров крупного очага в области базальных ядер справа, появление нового очага в правых отделах валика мозолистого тела (размером до 3,0×6,0 мм). Заключение: многоочаговое поражение обоих полушарий, червя мозжечка, моста, таламусов, мозолистого тела; глиома правого зрительного нерва; арахноидальные ликворные кисты малых размеров (полюс левой височной доли, четверохолмная и ретроцеребеллярная цистерны). Офтальмологический статус: в 2022 г. – гиперметропия слабой степени обоих глаз. Частичная атрофия зрительного нерва справа под вопросом; в 2023 г. диагностирован гиперметропический астигматизм обоих глаз; подтверждена глиома диска зрительного нерва правого глаза. Гамартом радужной оболочки глаз (узелки Лиша), а также изменений со стороны костной системы не выявлено.
По данным объективного осмотра: общее состояние удовлетворительное, рост 123 см, вес 20,5 кг (гипотрофия I степени). Подкожножировая клетчатка развита недостаточно. Лимфатические узлы не увеличены. Со стороны внутренних органов без патологии. На теле отмечаются множественные пятна (более 30) цвета «кофе с молоком» различной локализации и размеров. Отмечаются как мелкие множественные очаги (менее 0,5 см в диаметре), так и крупные гиперпигментированные зоны до 8 см. Нарушение речи. По данным генеалогического исследования наследственный анамнез не отягощен.
Был проведен поиск патогенных и вероятно патогенных вариантов, ассоциированных с направительным диагнозом, методом полногеномного секвенирования (пробанд, мать). По результатам полногеномного секвенирования (поиск однонуклеотидных вариантов и вариаций числа копий ДНК) выявлен ранее не описанный вариант нуклеотидной последовательности (chr17:31249069TCCTCAGAA>C, NM_001042492.3, c.4060_4068delinsC, p.Ser1354Leufs*23) в 30 из 58 экзонов гена NF1 в гетерозиготном состоянии, с глубиной покрытия 11x (рис. 1).
Согласно базе данных OMIM, патогенные варианты в гене NF1 приводят к НФ1 с аутосомнодоминантным типом наследования (#162200). В базе данных gnomADv4.1.0 вариант не встречается. В базе данных популяционных частот
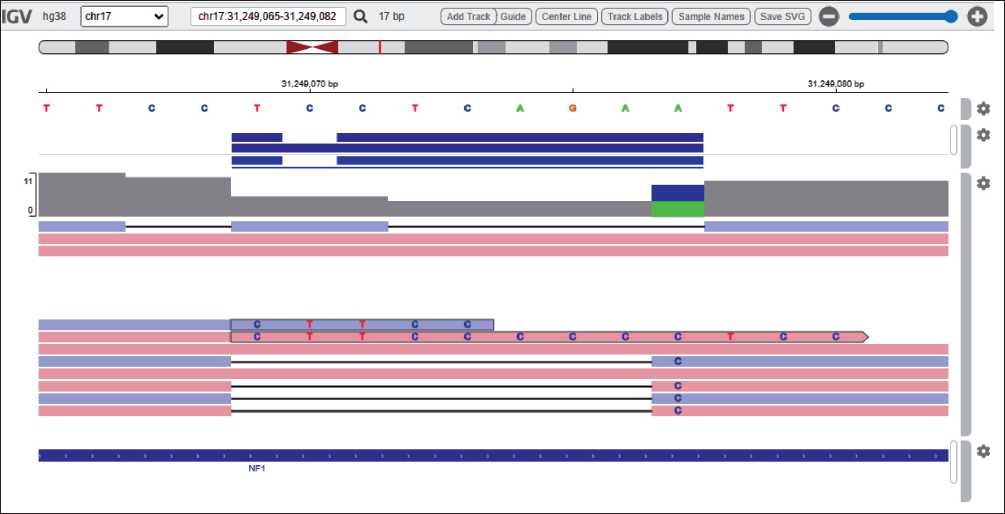
Рис. 1. NGS сиквенс пробанда (IGV browser). Примечание: рисунок выполнен авторами Fig. 1. Proband NGS sequencing (IGV browser). Note: created by the authors
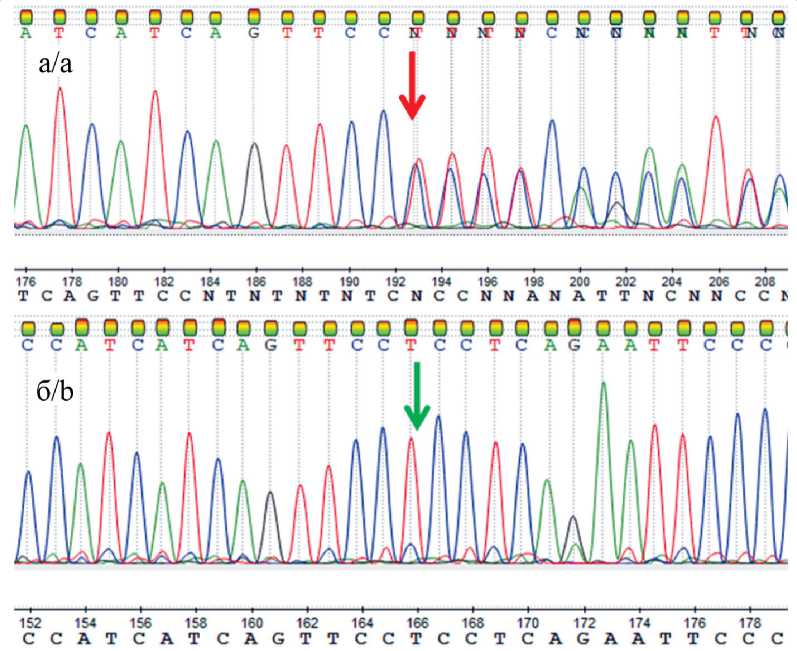
Рис. 2. Секвенирование по Сэнгеру у пробанда (а) и матери (б). Примечание: рисунок выполнен авторами Fig. 2. Sanger sequencing of proband (a) and proband’s mother (b). Note: created by the authors
генетических вариантов населения Российской Федерации данный вариант также не обнаружен. В базе данных ClinVar вариант не описан. По совокупности сведений выявленный вариант нуклеотидной последовательности следует расценивать как патогенный вариант, имеющий возможное отношение к фенотипу. Вариант подтвержден секвенированием по методу Сэнгера у пробанда в гетерозиготном состоянии и не выявлен у матери пробанда (рис. 2).
Обсуждение
Нейрофиброматоз I типа (neurofibromatosis type 1 #162200) вызывается гетерозиготными вариантами в гене нейрофибромина ( NF1 *613113), расположенного на хромосоме 17q11. Ген NF1 отвечает за синтез белка-онкосупрессора нейрофибромина, который преимущественно экспрессируется в клетках нейроэктодермального происхождения: нейронах, клетках Шванна (леммоцитах), олигодендроцитах и лейкоцитах. Нейрофибромин регулирует пролиферацию, миграцию, апоптоз, рост аксонов и развитие цитоскелета клетки через сигнальные пути Ras/MAPK, Akt/mTOR, ROCK/ LIMK/cofilin и cAMP/PKA. Более 80 % патогенных вариантов в гене NF1 приводят к синтезу нефункционального белка или полному отсутствию транскрипта [5]. Наиболее часто встречаются делеции всего гена NF1 , инсерции или делеции со сдвигом рамки считывания, а также варианты нарушения сплайсинга [6].
Золотым стандартом диагностики нейрофиброматоза является первоочередное исключение делеции гена NF1 (метод FISH, MLPA), а уже в дальнейшем, если патогенный вариант не выявлен, поиск более редких вариантов с использованием методов NGS (секвенирование гена, панели генов, полноэкзомное/полногеномное секвенирование). При этом полногеномное секвенирование позволяет идентифицировать как микроделеции/микро-дупликации (моногенные и полигенные), так и варианты в кодирующих и некодирующих частях генома, что позволит в дальнейшем ускорить диагностику более редких форм нейрофиброматоза.
