Клинико-эпидемиологическая характеристика популяции с болезнью Гоше в Челябинской области

Автор: Зуб Н.В., Жуковская Е.В.
Журнал: Человек. Спорт. Медицина @hsm-susu
Рубрика: Проблемы здравоохранения
Статья в выпуске: 6 (182), 2010 года.
Бесплатный доступ
Болезнь Гоше (БГ) - аутосомно-рецеесивное заболевание, обусловленное мутациями в структурном гене p-D-глюкоцереброзидазы, фермента, присутствующего в лизосомах всех типов тканей. Изменение структуры фермента в результате генных мутаций ведет к нарушению расщепления глюкоцереброзида, который накапливается в клетках всех тканей. При БГ выделяют три типа в зависимости от наличия неврологических признаков: ненейронопатический; острый нейронопатический; хронический нейронопатический. В исследовании, диагностике и лечении БГ последовательно реализовались все методические подходы, присущие наследственным болезням обмена. Они включают анализ патологических метаболитов, дефектного фермента, мутантного гена, методы ферментзаместительной терапии, субстратподавляющей и генотерапии.
Дети, болезнь гоше, методы ферментзаместительной терапии
Короткий адрес: https://sciup.org/147152811
IDR: 147152811
Clinik-epidemiological characteristic of the population with Gaucher disease in the Chelyabinsk area
Gaucher disease (GD) is an autosomal recessive condition caused by mutations in the P-D-glucoeerebrosidase structural gene; this enzyme is present in the lysosomes of all tissue types. Modification of the enzyme structure as a result of gene mutations leads to disorders in the glucocerebroside cleavage, and it is accumulated in the cells of all tissues. Three types of BG are distinguished: non-neuronopathic; acute neuronopathic; and chronic neuronopathic. All methodological approaches used in hereditary metabolic diseases were realized in the studies, diagnosis, and treatment of GD. These approaches include analysis of pathological metabolites, defective enzyme, mutant gene, and enzyme substitution, substrate suppressive, and gene therapy methods.
Текст научной статьи Клинико-эпидемиологическая характеристика популяции с болезнью Гоше в Челябинской области
Введение. Медицинская общественность ряда стран в 80-е годы минувшего столетия обратила внимание на редкие заболевания. Редкие или «орфанные» заболевания - это патология, которая встречается с незначительной частотой, жизнеугрожающая или хронически прогрессирующая, приводящая к смерти или инвалидности. В мировой практике разработаны количественные критерии редких заболеваний. В различных странах они варьируют от 1 на 1500-2500 человек населения страны.
В государствах Европейского союза (ЕС) насчитывается более 25 млн человек, страдающих разнообразными редкими заболеваниями, в России таких людей около 1,5 млн человек. В США, Японии, Сингапуре, Австралии, странах ЕС приняты законы, стимулирующие научные и производственные центры по разработке средств диагностики и лечения этих заболеваний [3].
Формулярным Комитетом Российской академии медицинских наук в декабре 2007 г. создана Профессиональная служба по редким заболеваниям, требующим дорогостоящего лечения, которая объединяет научных исследователей, врачей, менеджеров здравоохранения, провизоров и пациентов. Целью создания Профессиональной службы являлись общественный мониторинг за реализацией проекта по 7 редким нозологиям: гемофилия, муковисцидоз, гипофизарным нанизм, болезнь Гоше, миелолейкоз, рассеянный склероз после трансплантации органов и (или) тканей, оценка динамики качества жизни пациентов, их удовлетворенности ситуацией с лекарственным обеспечением и медицинской помощью в целом. В апреле 2008 г. результаты работы Профессиональной службы одобрены Президиумом РАМН. Было вынесено реше ние о подготовке проекта Государственной целевой программы по редким заболеваниям. Основные стратегические положения программы: централизация вопросов сложной диагностики в федеральных центрах и их филиалах, централизованное и персонифицированное лекарственное обеспечение, индивидуальный мониторинг, специальные методы регистрации медицинских технологий, открытость программы [2].
Наиболее изученной среди редких лизосомальных болезней накопления является болезнь Гоше (БГ) Это генетическое заболевание, обусловленное дефектом лизосомного фермента ^-D-глюкозидазы и характеризуется накоплением клеток Гоше в костном мозге, печени, селезенке, головном мозге и других органах. Впервые описание болезни Гоше опубликовано Р. Gaucher в 1882 г. [3, 4, 8]. Деление БГ на три типа основывается на присутствии (1 тип) или отсутствии (типы 2 и 3) неврологических симптомов. Клинические фенотипы встречаются с различной частотой. Самыми частыми является тип 1 БГ (частота 1:40000 до 1:60000 в разных популяциях), типы 2 и 3 встречаются реже (примерно 1:100000). Заболевание одинаково часто встречается у лиц обоего пола. Самая многочисленная когорта пациентов включает детей до 10 лет.
В качестве основных диагностических методик при БГ используются: измерение активности /TD-глюкозидазы в лейкоцитах; молекулярно-генетический анализ. Контроль за эффективностью проводимой терапии проводится по определению уровня хитотриазидазы в сыворотке.
Результаты молекулярно-генетического анализа в мире демонстрируют возможную концен-
Проблемы здравоохранения
трацию отдельных мутаций в различных этнических группах. Частота специфических мутантных аллелей варьирует в разных популяциях [1]. Наиболее частые мутации N370S, L444P, 84-85insG и IVS2+1G—>А составляют более 96 % мутантных аллелей у больных евреев ашкенази и менее 75 % мутантных аллелей у больных других национальностей. В Японии среди больных с болезнью Гоше не выявлено мутаций N370S и c.84-85ins G, а наиболее частыми являются мутации L444P и F213I [4, 8]. В португальской популяции больных мутация N370S составляет 63 % мутантных аллелей, часто встречаются 2 редкие мутации - G377S и N396T. В Швеции часто встречается 3 тип болезни Гоше, больные являются гомозиготами по точечным мутациям L444P.
Имиглюцераза показана для использования в качестве пожизненной ферментной заместительной терапии больных с подтвержденным диагнозом болезни Гоше типов 1 и 3, у которых имеются клинически значимые не-неврологические проявления болезни [5, 6].
Терапия имиглюцеразой оказывает такое же действие, как и природный энзим, представляя источник действующей глюкоцереброзидазы в клетки, где есть ее дефицит, и снижает накопление глюкоцереброзида. Под действием имиглюцеразы про исходит гидролиз гликолипида глюкоцеребразида до глюкозы и церамида по обычному пути метаболизма мембранных липидов. В связи с гетерогенностью болезни Гоше доза Церезима для каждого пациента должна подбираться индивидуально. Доза может повышаться или снижаться, в зависимости от успешности достижения клинических целей на основании оценки клинических проявлений [7, 8].
Цель работы: оценить клинические особенности пациентов с болезнью Гоше Челябинской области на фоне применения ферментозаместительной терапии.
Материалы и методы. В работу включено 11 пациентов с болезнью Гоше, зарегистрированных на территории Челябинской области; клинический метод (учитывая проспективный характер наблюдения в течение 30 лет, исследование носит продольный характер); биохимический метод исследования (измерение активности ^-D-глюкозидазы и хитотриозидазы); гистологический метод исследования.
Результаты обследования. Всего за 30-летний период наблюдения в детском онкогематологи-ческом центре г. Челябинска наблюдалось 11 детей. Средний возраст на момент диагностики 4 года. Клинический фенотип гетерогенен, но ни одного случая с нейропатическими проявлениями не заре-
Клинические особенности пациента с болезнью Гоше на фоне терапии
|
Дата |
Клинические особенности |
Паренхиматозные органы |
Показатели крови |
Костные изменения |
Хитотриа-зидаза (норма до 200 нМ/мг/ч) |
Лечение |
||
|
Печень, см |
Селезенка, см |
НЬ, г/л |
Тц, тыс. |
|||||
|
1985 г. (2 года) |
Диагноз установлен: в пунктате костного мозга клетки Гоше; гепатоспленомегалия; тромбоцитопения |
+ 4 |
+ 6 |
108 |
84 |
На рентгенографии в дистальных диафизах бедренных костей -широкие полосы просветления |
Не определялась |
Заместительная и симптоматическая терапия |
|
1990 г. (7 лет) |
Контрольное обследование в ГВЦ РАМН г. Москва. Диагноз болезни Гоше подтвержден; рентгенологические изменения бедренных костей |
+ 4 |
+ 6 |
80 |
75 |
На рентгенографии очаги деструкции в нижней трети обоих бедер |
Не определялась |
Заместительная и симптоматическая терапия |
|
1995 г. (12 лет) |
Сохранение гематоспленомегалии, анемии, тромбоцитопении |
+ 4 |
+ 6 |
82 |
72 |
Прежние |
Не определялась |
Заместительная и симптоматическая терапия |
|
2003 г. (20 лет) |
Нарастание анемии |
+ 4 |
+ 6 |
37 |
77 |
Прежние |
Не определялась |
Заместительная и симптоматическая терапия |
|
2007 г. (24 года) |
Положительная рентгенологическая динамика бедренных костей, определение уровня хи-тотриазидазы, назначение церезима в декабре |
+ 4 |
+ 7 |
88 |
78 |
На рентгенографии очаги деструкции в нижней трети обоих бедер не определяются |
13968 нМ/мг/ч |
Церезим из расчета 25 ед./кг х 2 раза в месяц |
|
2009 г. (26 лет) |
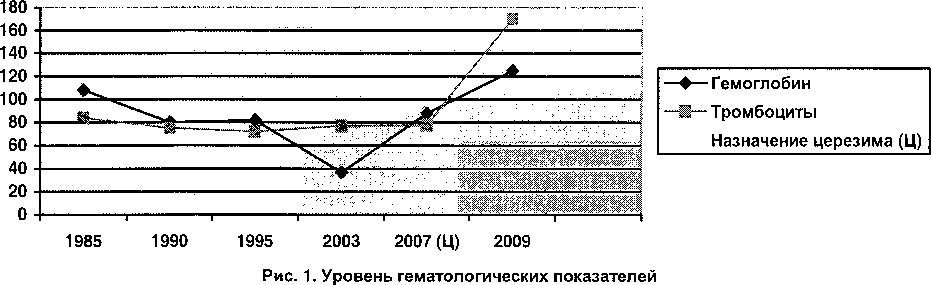
Улучшение гематологических показателей |
+ 4 |
+ 7 |
125 |
170 |
Нет |
Не определялась |
Церезим из расчета 25 ед./кг х 2 раза в месяц |
Зуб Н.В., Жуковская Е.В.
Клинико-эпидемиологическая характеристика популяции с болезнью Гоше...
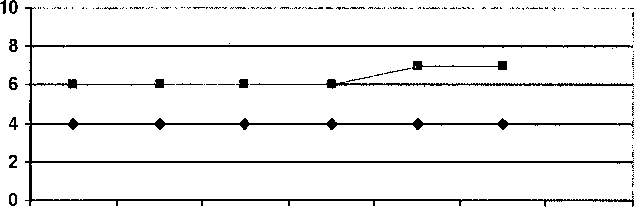
—♦—Печень
*—Селезенка
Назначение церезима(Ц)
1985 1990 1995 2003 2007(Ц) 2009
Рис. 2. Уровень паренхиматозных органов
гистрировано. Большинство пациентов было обследовано в медико-генетическом центре РАМН и им подтвержден 1 тип болезни Гоше. Частота этого наследственного заболевания 1:100000, что ниже общепринятых показателей почти на половину. Этот факт, как и отсутствие пациентов со 2 и 3 типами болезни Гоше также свидетельствует о существенных дефектах диагностики данной патологии. Учитывая высокий индекс межнациональной ассимиляции на территории Урала, не предоставляется возможным достоверно подтвердить принадлежность пациентов к этнической группе евреев ашкенази.
Клинический пример:
Пациент Е 75.2/01
В июле 1985 года обследован в Челябинской областной детской клинической больнице, выставлен диагноз: Болезнь Гоше 1 типа (см. таблицу). Диагноз подтвержден в НЦЗД РАМН в 1990 году.
Получал симптоматическую и заместительную терапии.
В ноябре 2007 года впервые проведено исследование активности ферментов /TD-глюкозидазы, которая составила 1,2 нМ/мг/ч (резко снижена), хитотриазидазы - 13968,0 нМ4МУФ/мл/ч (резко повышена). С 1 полугодия 2008 года проводится ферментная заместительная терапия препаратом Церезим в дозе 25 ед./кг массы тела внутривенно, капельно 1 раз в 2 недели. Аллергической реакции на препарат не отмечалось. Было выявлено, что применение Церезима привело к улучшению гематологических и костных показателей (рис. 1, 2). На фоне проводимой терапии показатели хитотриазидазы больше не определялись.
Таким образом, диагностика болезни Гоше требует проведения комплексного обследования, которое позволяет правильно установить диагноз, а значит назначить терапию на ранней стадии заболевания и улучшить качество жизни детей с данной патологией.
Список литературы Клинико-эпидемиологическая характеристика популяции с болезнью Гоше в Челябинской области
- Букина, Т.М. и др.//Вопросы гематологии, онкологии и иммунологии в педиатрии, 2004. -Т. 3, № 4. -С. 36-42.
- Воробьев, А.И. Руководство по гематологии/А.И. Воробьев, М.Д. Бриллиант. -М., 1985. -С. 157-351.
- Соколов, А.А. Редкие заболевания и сиротские медицинские технологии/А.А. Соколов. -СПб., 2008. -http://www.pharmvestnik>.
- Туре 1 Gaucher disease: Molecular, biochemical, and clinical characterization of patients from northern Portugal/O. Amaral, L. Lacerda, R. Santos et al.//Biochem Med Metab Biol, 1993. -V. 49.-P. 97-107.
- Mutations in Jewish patients with Gaucher disease/E. Beutler, T. Gelbart, W. Kuhl et al.//Blood, 1992. -V. 79. -P. 1662-1666.
- Eto, Y. Clinical and molecular characteristics of Japanese Gaucher disease/Y. Eto, H. Ida//Neurochem Res, 1999. -V. 24. -P. 207-211.
- Erikson, A. Neuronopathic forms of Gaucher's disease/A. Erikson//Presented at the First Workshop of European Working Group on Gaucher's Disease, October 13-16, 1994. -Trieste, Italy, 1994.
- Gaucher Registry Annual Report, 2004. -V. 5-10. -P. 4-12.
