Квантово-химическое моделирование эффективности использования полимеров в интерфейсных структурах

Автор: Калимуллина Луиза Раяновна, Лачинов Алексей Николаевич, Байбулова Галия Шафкатовна, Юсупов Азат Равилевич, Мухаммадамин Киан
Журнал: Математическая физика и компьютерное моделирование @mpcm-jvolsu
Рубрика: Моделирование, информатика и управление
Статья в выпуске: 2 т.23, 2020 года.
Бесплатный доступ
В данной работе был проведен квантово-химический анализ эффективности применения полиариленфталидов в интерфейсных структурах. Были проведены квантово-химические расчеты для молекулярных систем, представляющих собой модельные системы полимеров класса полиариленов - полиариленфталидов. Всего в рассмотрение входило 9 представителей данного класса соединений. Для всех молекул были проведены квантово-химические расчеты с использованием метода теории функционала плотности B3LYP/6-31+G(d) и теоретически оценены такие энергетические параметры, как полные энергии молекул и их отрицательных и положительных ионов в молекулярной и оптимизированной ионной геометриях; энергии занятых и вакантных молекулярных орбиталей; величины вертикального и адиабатического сродства к электрону и потенциала ионизации, а также дипольный момент.В настоящей работе предложен алгоритм обработки результатов квантово-химических расчетов на основании анализа энергетических характеристик мономерного звена полимера, позволяющий выявить определенную взаимосвязь между химической структурой органического соединения и электронными свойствами границы раздела металл/полимер. Предложенный алгоритм позволил выявить области максимального отклонения энергетических параметров и конкретные соединения, которые представляют интерес для формирования гетероструктур. Показана корреляция результатов, полученных с помощью методов квантово-химического моделирования с экспериментальными результатами по определению потенциальных барьеров на границе металл/полимер и проводимости вдоль интерфейса полимер/полимер.
Теория функционала плотности, гетероструктуры, органические диэлектрики, полиариленфталиды, боковая фталидная группа, сродство к электрону, потенциал ионизации, мономер
Короткий адрес: https://sciup.org/149129876
IDR: 149129876 | УДК: 544.03 | DOI: 10.15688/mpcm.jvolsu.2020.2.3
Quantum-chemical modeling of the efficiency of using polymers in interface structures
In this work, a quantum-chemical analysis of the effectiveness of the use of polyarylenephthalides in interface structures is carried out. Quantum chemical calculations are performed for molecular systems, which are model polymer systems of the polyarylene class - polyarylenephthalides. In total, 9 representatives of this class of compounds are considered. Quantum chemical calculations are performed for all molecules using the density functional theory method B3LYP/6-31 +G(d) and such energy parameters as the total energies of the molecules and their negative and positive ions in molecular and optimized ionic geometries; energies of occupied and vacant molecular orbitals; the values of vertical and adiabatic electron affinity and ionization potential, as well as the dipole moment are theoretically estimated.In this paper, the authors propose an algorithm for processing the results of quantum-chemical calculations based on the analysis of the energy characteristics of the polymer monomer unit, which makes it possible to reveal a certain relationship between the chemical structure of the organic compound and the electronic properties of the metal/polymer interface. The proposed algorithm makes it possible to identify areas of maximum deviation of energy parameters and specific compounds that are of interest for the formation of heterostructures. The correlation of the results obtained using the methods of quantum chemical modeling with the experimental results on the determination of potential barriers at the metal/polymer interface and conductivity along the polymer/polymer interface is shown.
Текст научной статьи Квантово-химическое моделирование эффективности использования полимеров в интерфейсных структурах
DOI:
Органические материалы (ОМ) и полимеры в их числе обладают чрезвычайно большим потенциалом, в качестве модельного одномерного объекта как для фундаментальных научных исследований, так и для практического применения не только в виде конструкционных материалов, но и в различных областях электроники. ОМ, как электронные материалы, отличает от неорганических больший уровень электрон-фононного взаимодействия. В связи с этим в гетероструктурах на границах раздела металл/ОМ можно наблюдать самосогласованные явления, приводящие к необычным с точки зрения одноэлектронной теории последствиям. По-видимому, одним из первых сообщений о наблюдении такого сорта явлений была работа [6], в которой обсуждались результаты исследования последовательного контактирования полимерной пленки с различными металлами. В этой работе на примере полимеров с большой шириной запрещенной зоны было установлено, что в результате контактирования полимерная пленка может накапливать объемный заряд, многократно превышающий тот, который предсказывает одноэлектронная теория, как результат выравнивания уровней Ферми. В этом смысле ОМ с большой шириной запрещенной зоны представляют собой удобный объект для тюнинга локализованных электронных состояний вблизи области контакта. Это заключение было подтверждено результатами большого количества различных экспериментов, проведенных с использованием субмикронных пленок различных несопряженных полимеров [4]. Основной результат всех этих работ можно определить как возможность управления электронными свойствами несопряженных (диэлектрических) ОМ в широком диапазоне электронных параметров (подвижность и концентрация носителей заряда, свойства потенциальных барьеров на границе раздела металл (полупроводник)/полимер, электропроводность) с помощью физически малых внешних воздействий [3]. Несмотря на большое количество работ, посвященных необычным электронным свойствам тонких пленок органических диэлектриков, до сих пор мало обсуждаются критериальные подходы к оценке той или иной органической химической структуры с точки зрения электронной эффективности ее использования в гетероструктуре, содержащей границу раздела металл (полупроводник)/полимер.
В связи с этим в настоящей работе предлагается рассмотреть алгоритм обработки результатов квантово-химических расчетов на основе анализа энергетических характеристик мономерного звена полимера, позволяющий выявить определенную взаимосвязь между химической структурой органического соединения и электронными свойствами границы раздела металл/полимер.
-
1. Материалы и методы исследования
-
2. Результаты квантово-химического моделирования
Среди несопряженных полимеров с уникальными электронными свойствами можно выделить группу кардовых полимеров – полиариленов. На примере полиариленфтали-дов выполнено наибольшее количество исследований аномальных электронных свойств гетероструктур типа металл/полимер/металл. В тонких пленках этих полимеров при инжекции носителей заряда из электродов было обнаружено множество эффектов, связанных с изменением проводимости от диэлектрика до металла. В настоящей работе была рассмотрена группа полиариленфталидов, так как для соединений этого класса имеется наибольшее количество экспериментальных результатов. Для проведения квантово-химических расчетов использовался метод теории функционала плотности в приближении B3LYP/6-31+G(d).
В таблице 1 представлены оптимизированные геометрические структуры 9 мономерных соединений из группы полиариленфталидов. Функциональной группировкой является фталидная группировка.
Мы определяем функциональными группами те, которые в полимерной структуре являются боковыми группами. Безусловно, этот выбор субъективен, но он связан с рядом известных свойств данных соединений. Ранее [5] отмечалось, что молекулярные орбитали боковой фталидной группы, например, полидифениленфталида, участвуют в формировании нижней вакантной орбитали полимера. А взаимодействие этой группы с избыточным электроном приводит к возникновению глубоких электронных состояний в запрещенной зоне полимера. Верхняя занятая молекулярная орбиталь сформирована в этом соединении электронными состояниями скелетной части макромолекулы.
Таблица 1
Оптимизированные геометрические структуры девяти мономерных молекул из группы полиариленфталидов
|
№ п/п |
Брутто-формула |
Изображение молекулы |
|
1 |
С 20 Н 14 SO 2 |
^ |
|
2 |
С 20 Н 12 SO 2 |
I * > |
|
3 |
С 26 Н 18 O 2 |
t * * X v *# = -* ,‘е-^ Л'А ^А |
|
4 |
С 21 NH 15 O 2 |
ч * ^yVv ^^^ к е~- /МАб./>е^ |
|
5 |
С 20 Н 14 О з |
С Сс %; с, С |
|
6 |
С 20 Н 12 О з |
*ТгА>чА А* |
|
7 |
С 20 NH 13 О 2 |
’ Л ; |
|
8 |
С 21 Н 14 О 2 |
|
|
9 |
С 20 Н 14 О 2 |
Для всех молекул были проведены квантово-химические расчеты и теоретически оценены следующие энергетические параметры: полная энергия Е 0 , энергия нижней вакантной молекулярной орбитали E LUMO , энергия верхней занятой молекулярной орбитали E HOMO , вертикальное сродство к электрону EA v , адиабатическое сродство к электрону EA a , вертикальный потенциал ионизации IP v , адиабатический потенциал ионизации IP a , а также дипольный момент D . Выбор энергетических параметров не является случайным. Ширину расщепления между верхней занятой и нижней вакантной молекулярной орбиталью можно ассоциировать с шириной запрещенной зоны молекулы. А дипольный момент молекулы оказывает определяющую роль в формировании гетероструктур на основе полимеров класса полиариленов [1]. В таблице 2 представлены результаты полученных расчетов для мономеров из группы полиариленфталидов.
Таблица 2
Оптимизированные геометрические структуры девяти мономерных молекул из группы полиариленфталидов
|
№ п/п |
Брутто-формула |
E o , Хартри |
- E lumo, эВ |
- E homo, эВ |
EA v , эВ |
EA a , эВ |
IP v , эВ |
IP a , эВ |
D, Д |
|
1 |
C 20 H 14 SO 2 |
- 1318 , 601 |
1 , 34 |
5 , 79 |
0 , 21 |
0 , 54 |
7 , 38 |
7 , 18 |
5 , 00 |
|
2 |
C 20 H 12 SO 2 |
- 1317 , 424 |
1 , 38 |
6 , 86 |
0 , 09 |
0 , 45 |
7 , 48 |
7 , 51 |
5 , 48 |
|
3 |
C 26 H 18 O 2 |
- 1151 , 396 |
1 , 32 |
5 , 88 |
- 0 , 10 |
0 , 32 |
7 , 23 |
7 , 04 |
5 , 10 |
|
4 |
C 21 N H 15 O 2 |
- 1013 , 905 |
1 , 28 |
5 , 41 |
0 , 24 |
0 , 56 |
6 , 95 |
6 , 83 |
3 , 21 |
|
5 |
C 20 H 14 O 3 |
- 995 , 660 |
1 , 23 |
6 , 16 |
0 , 37 |
0 , 82 |
7 , 77 |
7 , 86 |
5 , 92 |
|
6 |
C 20 H 12 O 3 |
- 994 , 478 |
1 , 36 |
6 , 06 |
0 , 11 |
0 , 30 |
7 , 65 |
7 , 57 |
5 , 37 |
|
7 |
C 20 NH 13 O 2 |
- 974 , 621 |
1 , 29 |
5 , 52 |
0 , 26 |
0 , 44 |
7 , 13 |
7 , 15 |
3 , 26 |
|
8 |
C 21 H 14 O 2 |
- 958 , 574 |
1 , 32 |
5 , 83 |
0 , 21 |
0 , 54 |
7 , 40 |
7 , 15 |
4 , 30 |
|
9 |
C 20 H 14 O 2 |
- 920 , 484 |
1 , 31 |
6 , 18 |
0 , 17 |
0 , 53 |
7 , 71 |
7 , 50 |
5 , 05 |
Детальное сравнение параметров для девяти рассмотренных молекул группы по-лиариленфталидов позволило выявить некоторые особенности. В частности, минимальное значение потенциала ионизации (как адиабатического, так и вертикального) соответствует молекуле, содержащей атом азота в основной цепи. А максимальный потенциал ионизации (адиабатический и вертикальный) наблюдается для молекулы с атомом кислорода в структуре основной цепи (полидифениленоксидфталид). В группе полиариленфталидов присутствует одно соединение с отрицательным значением вертикального сродства к электрону (молекула № 3 — политерфениленфталид). В работе [2] были исследованы некоторые представители полимеров класса полиариленфтали-дов (полидифениленсульфидфталид, политерфениленфталид, полидифениленоксидфта-лид, полидифениленфталид) и экспериментально определены величины потенциальных барьеров на границе металл/полимер. Сопоставление результатов, полученных в итоге квантово-химических расчетов, с результатами, полученными в работе [2], позволяет утверждать, что величина потенциального барьера коррелирует с потенциалом ионизации и шириной запрещенной зоны, оцененной как разность энергий верхней занятой и нижней вакантной молекулярной орбитали.
Значение дипольного момента максимально для полидифениленоксидфталида (5,92 Д), но минимально для молекулы, содержащей атом азота в основной цепи (молекула № 4), и составляет 3,21 Д. В работе [1] экспериментально было показано, что наибольшее значение проводимости наблюдается вдоль интерфейса полидифениленоксид-фталида, наименьшее для политерфениленфталида, что согласуется с представленной в данной работе моделью. Полимеры с большим значением дипольного момента (по-лидифениленоксидфталид) способны к спонтанному дипольному упорядочению боковых функциональных групп полимерных молекул.
Форма представления в виде таблицы 2 затрудняет анализ из-за большого количества присутствующих в ней энергетических параметров и отсутствия явных тенденций по изменению каждой из характеристик. Кроме того, представление электронных параметров в таком виде не позволяет выявить закономерности в изменении параметров и провести сравнение этих соединений с точки зрения их возможного использования в гетероструктурах. Поэтому была необходима дополнительная математическая обработка полученных данных. Все рассчитанные параметры были выстроены в порядке возрастания полной энергии. Затем для каждого рассчитанного параметра находилось максимальное значение, и все другие значения данного параметра нормировались на это максимальное значение. Аналогично были пронормированы все значения полной энергии молекул. Далее уже с нормированными параметрами были построены графики зависимости всех величин от полной энергии. На рисунке 1 представлен полученный график для группы полиариленфталидов.
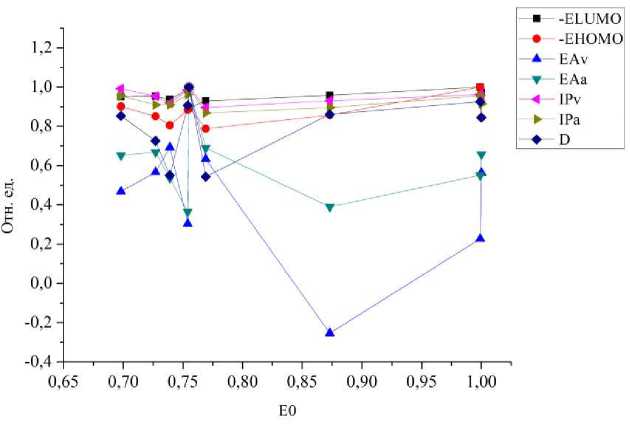
Рис. 1. График зависимости рассчитанных параметров от полной энергии для полиариленфталидов
На полученном графике можно выделить две основные области: область малых энергий (до 0,75) и область высоких энергий (от 0,75 до 1). В первой области для всех молекул по всем параметрам наблюдаются осцилляции. Ряд параметров меняется слабо (-ELUMO, -EHOMO, IPv, IPa). В то же время наибольшие изменения наблюдаются для трех параметров (EAv, EAa, D), то есть они, по-видимому, являются наиболее структурно зависимыми. Наличие одного соединения с отрицательным вертикальным сродством к электрону создает существенный разброс значений по данному параметру. Наименьшее значение дипольного момента в группе полиариленфталидов составляет 3,21 Д, а наибольшее значение равно 5,92 Д (отличаются в 1,84 раза). По-видимому, именно адиабатическое сродство к электрону, вертикальное сродство к электрону и дипольный момент являются наиболее чувствительными к химической структуре параметрами. Поскольку наибольший интерес представляла именно область изменений, то для уточнения данного вопроса был продифференцирован график, изображенный на рисунке 1. В результате проведенной процедуры дифференцирования был получен новый график, приведенный на рисунке 2.
Процедура дифференцирования необходима была для того, чтобы убрать лишние и выделить основные параметры и структуры. Как было отмечено ранее, наиболее чувствительными параметрами, сильно зависящими от структуры молекулы, являются вертикальное сродство, адиабатическое сродство и дипольный момент.
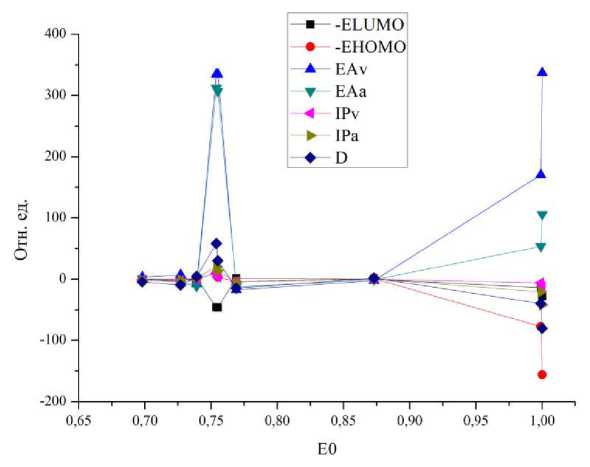
Рис. 2. График зависимости первой производной рассчитанных параметров от полной энергии для полиариленфталидов
На основании графика, изображенного на рисунке 2, можно утверждать, что в группе полиариленфталидов соединениями с высокой возможностью управления энергетическими параметрами являются следующие: молекула № 1 (полидифениленсульфидфта-лид) и № 4 и № 5 (полидифениленоксидфталид), так как именно для этих соединений наблюдаются осцилляции энергетических параметров. Очевидно, что адекватность предложенного подхода необходимо проверить в реальных экспериментах на более широком ряду полимеров, например, включающем три группы соединений: полиариленфталиды, полиариленсульфофталиды и полиариленфталимидины.
Список литературы Квантово-химическое моделирование эффективности использования полимеров в интерфейсных структурах
- Влияние дипольного упорядочения на электрофизические свойства границы раздела двух органических диэлектриков / Р. М. Гадиев, А. Н. Лачинов, А. Ф. Галиев, Л. Р. Калимуллина, И. Р. Набиуллин // Письма в Журнал экспериментальной и теоретической физики. - 2014. - Т. 100, № 4. - C. 276-280.
- Исследование транспорта носителей заряда через границу металл - полимер класса полиариленфталидов / А. Р. Юсупов, Р. Г. Рахмеев, А. Н. Лачинов, Л. Р. Калимуллина, А. С. Накаряков, А. А. Бунаков // Физика твердого тела. - 2013. - Т. 55, № 7. - C. 1392-1395.
- Лачинов, А. Н. Электроника тонких слоев широкозонных полимеров / А. Н. Лачинов, Н. В. Воробьева // Письма в Журнал экспериментальной и теоретической физики. - 2006. - Т. 176, № 12. - C. 1249-1266.
- О возможном механизме аномально высокой проводимости тонких пленок диэлектриков / Ю. А. Берлин, С. И. Бешенко, В. А. Жорин, А. А. Овчинников, Н. С. Ениколопян // ДАН СССР, Сер. Физика. Химия. - 1981. - Т. 260, № 6. - C. 1386-1390.
- A theoretical study of the chemical structure of the non-conjugated polymer poly(3, 3'-phthalidylidene-4, 4'-biphenylene) / N. Johansson, A. N. Lachinov, S. Stafstro¨m, T. Kugler, W. R. Salaneck // Synthetic Metals. - 1994. - Vol. 67, № 1. - P. 319-322.
- Duke, C. B. Charge-Induced Relaxation in Polymers / C. B. Duke, T. J. Fabish // Physical Review Letters. - 1976. - Vol. 37, № 16. - P. 1075-1078.
