Масштабируемость квантово-химических расчетов кристаллических материалов на суперкомпьютере «Торнадо ЮУрГУ»

Автор: Юшина Ирина Дмитриевна, Матвейчук Юрий Васильевич, Барташевич Екатерина Владимировна
Статья в выпуске: 3 т.11, 2022 года.
Бесплатный доступ
В работе обсуждаются рекомендации по рациональному использованию вычислительных ресурсов суперкомпьютера "Торнадо ЮУрГУ" для решения квантовохимических расчетных задач c учетом периодических граничных условий в программе CRYSTAL17. Решение таких задач необходимо для моделирования структуры и свойств кристаллов и, углеродных материалов. Проанализированы данные о временных характеристиках квантовохимических расчетов материалов различного состава и структуры, как для простых систем, таких, как трехмерный силицированный графит, так и для двумерных поверхностей углеродных материалов с диглицидиловым эфиром бисфенола A в роли сорбированной молекулы. Определены различия в масштабируемости расчетов в зависимости от их типа, размера периодически повторяющегося фрагмента структуры (элементарной ячейки), ее симметрии и входных параметров многоэлектронной системы, моделируемой на основе теории функционала электронной плотности в различных приближениях. Установлены рекомендуемые оптимальные параметры для различных типов химических и материаловедческих задач, решаемых в целях разработки цифровых двойников материалов. Обнаружено, что для относительно больших систем критическим является существенное увеличение требующегося объема временных файлов с увеличением числа используемых узлов, что приводит к неоптимальному режиму расчетов и возможному сбою. Показано, что масштабируемость расчетов колебательных характеристик кристаллов существенно ниже, чем для расчетов, направленных на поиск наиболее энергетически выгодной структуры кристалла, независимо от числа атомов в элементарной ячейке вычисляемой структуры.
Цифровой двойник материалов, квантово-химические расчеты, функциональные материалы, оптимизация вычислений, масштабируемость
Короткий адрес: https://sciup.org/147238352
IDR: 147238352 | УДК: 541.65/.654, | DOI: 10.14529/cmse220304
Scalability of quantum chemical calculations of crystalline materials using “Tornado” supercomputer in South Ural State University
Technical results of multiscale modeling of crystalline materials of different structure and composition has been presented and discussed. The scalability of different types of quantum chemical calculations using resources of "Tornado" supercomputer has been studied. Such tasks are extremely relevant in the field of modeling of structure and properties of crystals and carbon materials. The range of scalability has been reported for the systems of different size, composition, symmetry and level of modeling in the framework of density functional theory with atomic basis sets. A list of recommendations has been formulated presenting the optimal parameters for different types of material science tasks on the way to digital twin design. Analysis of calculation time for different systems and calculation types has been performed. A list of simple systems such as 3D silicon-substituted graphite as well as complex systems consisting of carbon surfaces (nanotube and graphene layer) and Bisphenol A diglycidyl ether as molecule on a surface. It is revealed that for large systems the most critical condition is the increase of the size of temporary files with the increase of number of nodes leading to possible failure of the calculation. It was shown that the scalability of the calculations of vibration properties of crystals is significantly lower than of the search of energetically preferable structure no matter how many atoms are located in the elementary part of the cell of computed structure.
Текст научной статьи Масштабируемость квантово-химических расчетов кристаллических материалов на суперкомпьютере «Торнадо ЮУрГУ»
Структурные модели химических соединений и материалов представляют собой, как правило, сложные равновесные атомно-молекулярные системы, в которых, в зависимости от уровня модели, учитывается распределение электронной плотности, конкретизированы позиции атомов, установлены типы химических связей. Точная оценка количественных взаимосвязей «структура – свойство» тесно связана с проблемой накопления и хранения химической информации, которая в перспективе позволит разрабатывать и внедрять технологии, связанные с использованием цифровых двойников химических соединений и материалов, что относится к задачам, отвечающая вызовам нашего времени. Цифровые двойники позволяют моделировать внедрение новых материалов, выявлять экономические и экологические риски при использовании инноваций еще на этапе проектирования. Отметим, что
Масштабируемость квантовохимических расчетов кристаллических материалов ... оптимальный выбор материала при проектировании продукта является важнейшим вопросом при реализации устойчивого производства [1] .
На протяжении многих лет отечественными и зарубежными исследователями проводились квантовохимические расчеты структуры и свойств химических структур с различной периодичностью для задач катализа [2] , материаловедения [3] , оптики и биологической активности [4] . Одной из отличительных особенностей этого круга задач в периодическом приближении является их возрастающая потребность в объеме вычислительных мощностей и большие временные затраты, необходимые для получения достоверных результатов. Одним из передовых программных продуктов для решения квантовохимических задач является CRYSTAL17, разрабатываемый в университете г. Турина [4] . Методические аспекты использования этой программы хорошо освещены в отечественной литературе в работах Р.А. Эварестова [5] . Однако вопросы оптимизации расчетов в условиях ограниченных вычислительных и временных ресурсов обычно остаются за рамками научных исследований, в то время как такие оценки необходимы для результативного использования имеющихся ресурсов и достижения результатов в условиях временных ограничений.
Интуитивный анализ более чем десятилетнего опыта расчетов в программе CRYSTAL17 на суперкомпьютере «Торнадо ЮУрГУ» показывает, что пользователи, как правило, не используют все имеющиеся возможности суперкомпьютера для минимизации компьютерного времени расчетов. Так, например, при работе с большей частью квантово-химического программного обеспечения редко обращается внимание на возможности существенного сокращения используемого объема дисковой памяти и обмена с этой памятью, что является в подавляющем большинстве случаев лимитирующим фактором времени расчетов. Как правило, работа с оперативной памятью является более предпочтительной в смысле временных затрат для очень многих прикладных расчетов, где не используются большие объемы промежуточных данных. Из этого правила есть исключения, например, расчет колебательных характеристик молекул или кристаллов с большим числом атомов в элементарной ячейке, когда установленной оперативной памяти недостаточно, и без большого количества дисковой памяти и интенсивного обмена с ней не обойтись. Необходимость систематического анализа возможностей ускорения решения задач с использованием квантовохимического программного обеспечения не теряет своей актуальности. Одной из таких возможностей является корректный выбор количества узлов суперкомпьютера, используемых для каждой отправляемой на расчет задачи, в зависимости от типа задачи, необходимых параметров сходимости расчета и величины рассчитываемых объектов. Для выяснения информации, позволяющей осуществлять правильный выбор при отправке задач на расчет, нами поставлены следующие задачи:
\bullet определить возможность масштабирования вычислительных задач разных типов: оптимизации периодических 1D, 2D и 3D структур, решение спектральной задачи для 3D структур в CRYSTAL17 [6] ;
\bullet определить оптимальные количества узлов, необходимых для оптимальных временных затрат с учетом загрузки внутренней сети и дисковой памяти для химических объектов разных размеров.
-
1. Вычислительные эксперименты
-
2. Результаты и их обсуждение
В соответствии с этими задачами нами были выбраны следующие объекты исследования: 2D поверхность, состоящая из трехатомного слоя частично замещенного кремнием графита (силицированный графит) (1); 3D кристалл силицированного графита (2); бисфенол A, сорбированный на графеновом листе (3); бисфенола A, сорбированный на углеродной нанотрубке (УНТ) (4); кристаллическая структура каркасного соединения COF-102 (5) [7]. Основное тестирование осуществлялось для поиска равновесной геометрии системы. С одной стороны, это обязательная процедура моделирования, с другой стороны, для такого типа задач имеются статистические данные по эффективности распараллеливания алгоритма вычислений, предоставленные разработчиками программы [4]. Параметры моделирования для каждого объекта представлены в табл. 1, где № АО обозначено число атомных орбиталей на ячейку, а число шагов соответствует числу циклов на пути к найденному минимуму энергии. Графические результаты изменения времени расчета представлены на рис. 1–4.
Таблица 1. Структурные параметры объектов исследования и параметры расчетов
|
Объект |
Функционал |
№ АО |
Число атомов |
Симметрия |
Число шагов |
|
1 |
HSE06 |
116 |
8 |
P63/mmc |
59 |
|
2 |
HSE06 |
344 |
24 |
P1 |
61 |
|
3 |
PBE0 |
3830 |
289 |
P1 |
75 |
|
4 |
PBE0 |
3606 |
273 |
P1 |
91 |
|
5 |
B3LYP |
3252 |
13 |
I-43d |
41 |
Формирование набора одновременно выполняемых задач осуществлялось в динамическом режиме в зависимости от получаемых результатов в текущем времени. Для задач поиска равновесной геометрии всех объектов был осуществлен предварительный подбор метода и параметров расчета для нахождения оптимизированной структуры. В дальнейшем одна и та же задача была стартована на разном числе узлов с контролем объема временных файлов на этапе оптимизации геометрии и прохождения 5 шагов. Диапазон узлов на один расчет выбирался от 1 до 140. Задачи расчета колебательных спектров проводились на небольшом числе узлов, так как ранее в стандартном режиме работы суперкомпьютера была неоднократно замечена вероятность срывов параллельно выполняемых задач из-за перегрузки сети при данном типе расчетов. В данном случае был протестирован диапазон от 1 до 4 узлов на 1 расчет [8] .
Таблица 2. Характеристики кластера Торнадо ЮУрГУ
|
Характеристика |
Значение |
|
Количество процессорных узлов |
480 |
|
Тип процессора |
Intel Xeon X5680 (Gulftown, 6 ядер по 3.33 ГГц) |
|
Оперативная память |
24 Гб (DDR3-1333) |
|
Операционная система |
Linux CentOS |
|
Соединительная сеть |
InfiniBand QDR (40 Гбит/с) |
В соответствии с данными, представленными на рис. 1–4, видно, что объем временных файлов растет линейно в зависимости от числа узлов для одной и той же задачи, независимо от объекта вычисления. Объем файлов также зависит от количества атомов в системе: от десятков мегабайт для 3D силицированного графита с восемью атомами в ячейке, до 11 Гб для 2D комплекса бисфенола А с графеном с 273 атомами в ячейке. Следует отметить, что среди выбранных для исследования химических соединений со структурой, имеющей различную размерность — 2D и 3D, можно выделить хорошо масштабируемые: 2D поверхность силицированного графита и плохо масшабируемые: кристалл COF-102. Для количественной оценки времени расчета в зависимости от числа используемых узлов (табл. 3) было проведено сравнение вычислений при увеличении числа узлов в 10 раз в зависимости от их исходного числа: 1 и 10 узлов, 2 и 20 и так далее. Из табл. 3 мы видим, что для плохо масштабируемых вычислений показатель ускорения едва превышает единицу для любого соотношения начального и конечного числа узлов.
Таблица 3. Показатели ускорения расчета при использовании различного количества узлов и различных объектов 1–5
|
Отношение числа узлов |
1 |
2 |
3 |
4 |
5 |
|
1/10 |
6.3 |
8.7 |
1.7 |
1.7 |
1.5 |
|
2/20 |
3.1 |
7.9 |
1.3 |
1.5 |
1.2 |
|
4/40 |
2.8 |
6.9 |
1.1 |
1.2 |
1.0 |
|
10/100 |
2.0 |
4.8 |
– |
– |
– |
|
1/2 |
1.8 |
1.95 |
1.3 |
1.1 |
1.3 |
Однако для силицированного графита, как для 2D, так и 3D размерностей структуры, наблюдается существенный выигрыш во времени, хотя он значительно снижается при увеличении исходного числа узлов. Кроме того, для 2D варианта было протестировано самое большое число узлов на одну задачу — 140. Даже в этом случае наблюдалось небольшое сокращение времени расчета, хотя и несоизмеримое с увеличением используемых вычислительных мощностей (рис. 2) . Для остальных рассмотренных объектов (рис. 1, 3, 4) выигрыш во времени расчета при увеличении вычислительных мощностей неоправданно мал. При этом очень быстро достигается пороговое значение числа узлов, когда дальнейшее увеличение этого числа вызывает рост времени расчета.
Таблица 4. Результаты расчета колебательных характеристик кристаллов силицированного графита (1) и COF-102 (5)
|
Параметр |
1 |
1 |
1 |
1 |
5 |
5 |
5 |
|
Число узлов |
1 |
2 |
4 |
8 |
1 |
2 |
4 |
|
Время расчета, мин |
130 |
79 |
61 |
43 |
362 |
439 |
311 |
|
Объем временных файлов, Мб |
213 |
245 |
289 |
326 |
12700 |
18000 |
19800 |
Оценка оптимального режима вычислений колебательных характеристик является более сложной задачей. В силу особенностей алгоритма такой тип задачи включает вычисление изменения энергии и вторых частных производных для элементов матрицы расстояний при смещении каждого симметрически-независимого атома по всем трем направлениям. В связи с этим решение спектральной задачи требует намного больших временных затрат. Тестирование решения спектральной задачи осуществлялось на кристаллических высокосимметричных объектах. В соответствии с полученными данными отметим, что для кристаллов с небольшим числом атомов в ячейке (cилицированный графит 2D) и для крупной ячейки кристаллического каркаса COF-102 наблюдалась в целом аналогичная картина, что и в задаче поиска равновесной геометрии. Однако если для COF-102 оптимизация геометрии являлась практически не масштабируемым процессом, то для расчета колебательных характеристик выигрыш во времени при увеличении числа узлов оказывается более существенным.
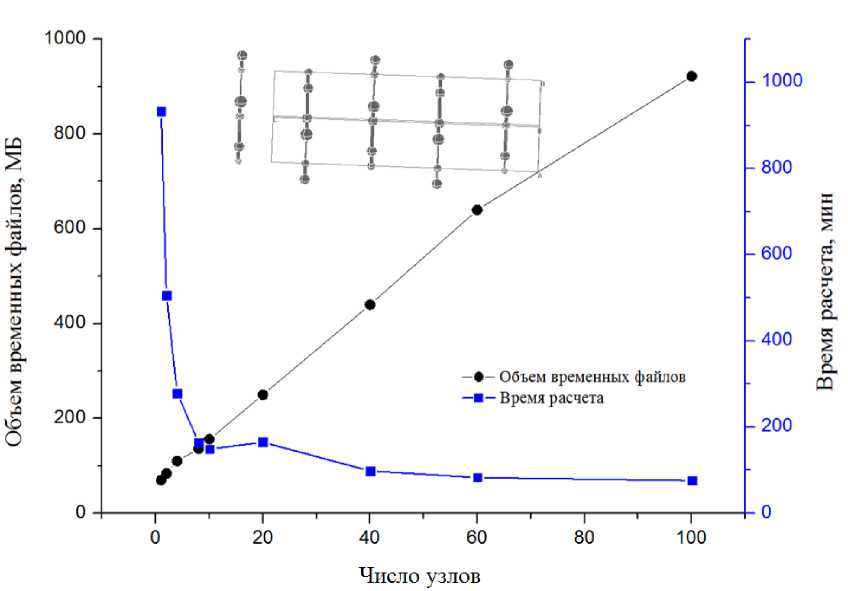
Рис. 1. Результаты расчетов кристалла силицированного графита (3D, 8 независимых атомов в ячейке, высокая симметрия)
Анализ объема временных файлов показывает, что эта величина для решения спектральной задачи оказывается существенно большей, чем для задачи оптимизации геометрии, хотя размер файлов растет в зависимости от числа узлов (табл. 4) . Это связано с особенностями алгоритма расчета, который включает два последовательно идущих вычисления для каждого смещения атомов: вычисление фазы Бэрри и работа с временными файлами. Если первый процесс сопоставим с вычислительными затратами при оптимизации геометрии и способен масштабироваться, то второй характеризуется высокой интенсивностью обмена данными между оперативной и дисковой памятью. Поэтому увеличение выделенного числа узлов для расчета частот колебаний высокосимметричных структур с большим количеством атомов в ячейке (> 15–20) скорее всего будет замедлять общее время расчета из-за пропорционально возрастающего обмена данными с файловым хранилищем и высокой загрузки внутренней сети суперкомпьютера. То есть, увеличение числа выделенных узлов для решения спектральной задачи и расчета колебательных спектров кристаллов имеет смысл только для сравнительно небольших объектов с числом атомов в ячейке не более 10–15. Небольшой выигрыш во времени расчета больших структур сопряжен с риском сбоя выполнения этой и других задач на суперкомпьютере.
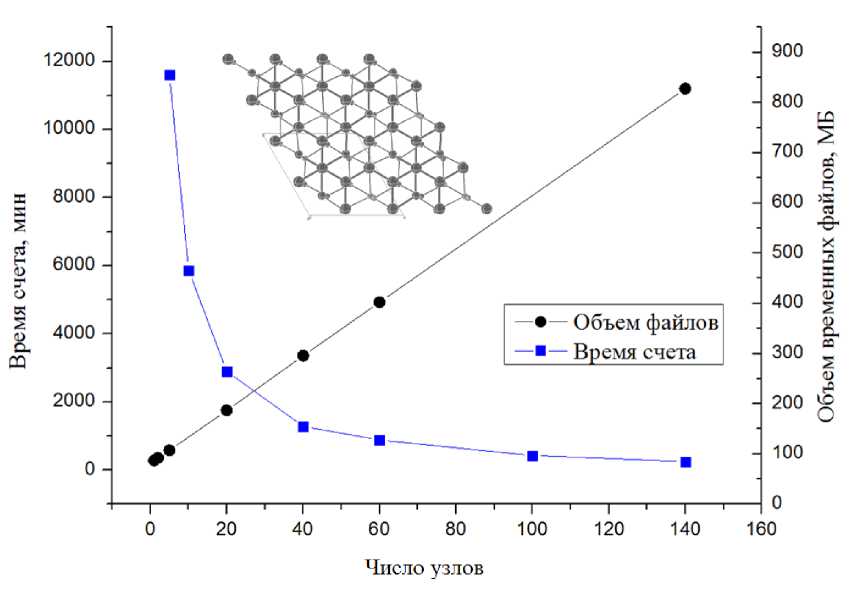
Рис. 2. Результаты расчетов слоя силицированного графита (2D, 24 независимых атома в ячейке, отсутствие симметрии)
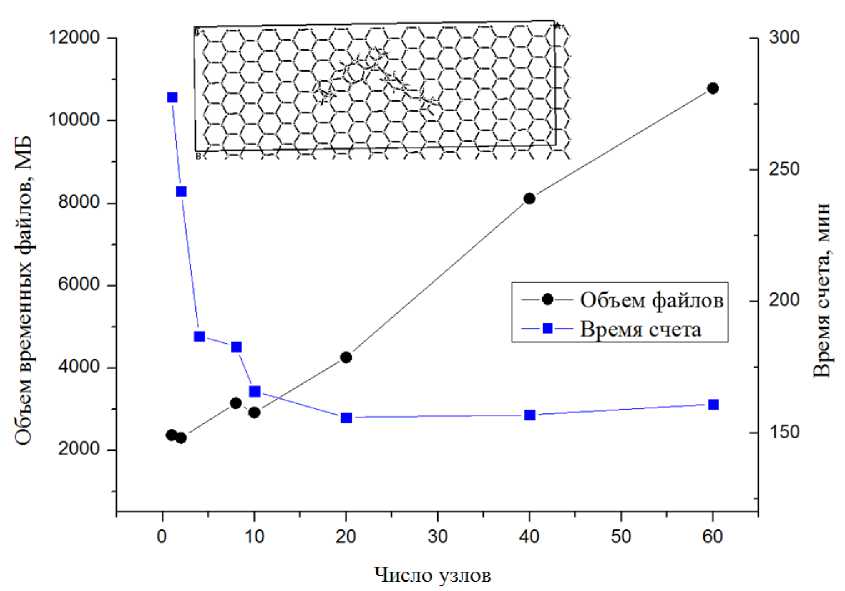
Рис. 3. Результаты расчетов комплекса бисфенола А, сорбированного на поверхности графена (2D, 273 атома в ячейке, низкая симметрия)
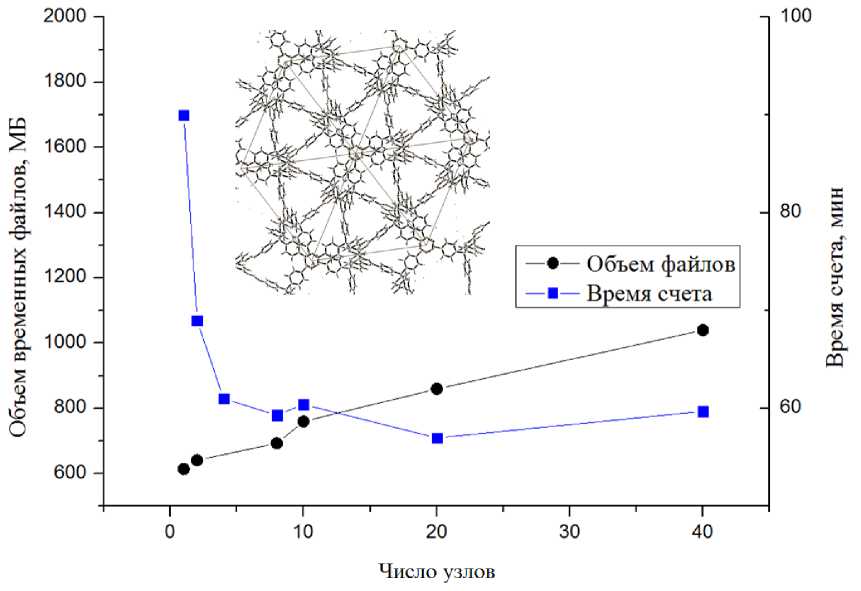
Рис. 4. Результаты расчетов кристалла ковалентного каркаса COF-102 (3D, 13 независимых атомов в ячейке, высокая симметрия)
Заключение
Таким образом, по результатам анализа временных затрат для решения различных квантовохимических задач, а именно: при поиске наиболее энергетически выгодной структуры кристалла или слоя и при расчете колебательных характеристик периодических структур на примере материалов с различным количеством атомов в элементарной ячейке, были выявлены следующие особенности и перспективы масштабируемости задач. Наибольшей масштабируемостью характеризуется оптимизация координат атомов в ячейке cили-цированного графита 2D, полученного из кристалла с высокой симметрией, в то время, как расчеты комплекса диглицидилового эфира бисфенола А на поверхности 2D графена за счет большого числа атомов в ячейке характеризуются большим объемом временных файлов и обладают существенно меньшими возможностями масштабирования. Структура кристаллического каркаса COF-102, с одной стороны, характеризуется высокой симметрией, а с другой — большим числом атомов в элементарной ячейке, и при увеличении числа узлов практически не демонстрирует выигрыша во времени расчета в процедуре оптимизации геометрии. Количество независимых атомов в элементарной ячейке исследуемой структуры материала влияет на размер временных файлов в ходе расчета, а время расчетов, в свою очередь, увеличивается с ростом числа используемых узлов. В связи с этим, для систем с большим количеством атомов, приходящихся на одну элементарную ячейку, или при использовании расширенных наборов атомных орбиталей – числа функций в базисном наборе, существенным оказывается увеличение объема временных файлов с увеличением числа используемых узлов, которое приводит к неоптимальному режиму расчетов и возможному сбою. Выяснено, что масштабируемость расчетов колебательных характеристик молекулярных кристаллов, в частности, крупных кристаллических каркасов, существенно ниже, чем для расчетов, направленных на поиск наиболее энергетически выгодной структуры, независимо от числа атомов в элементарной ячейке вычисляемой системы. Вследствие этого, для систем с большим числом атомов в ячейке и долгим временем расчета оказывается нерациональным увеличение числа используемых узлов больше четырех в связи с удлинением временного интервала активной работы с файловым хранилищем при записи временных файлов и связанным с этим увеличением вероятности сбоя параллельно идущих расчетов.
Работа выполнена при финансовой поддержке поддержке гранта РНФ № 22-13-00170.
Список литературы Масштабируемость квантово-химических расчетов кристаллических материалов на суперкомпьютере «Торнадо ЮУрГУ»
- Xiang F., Zhang Z., Zuo Y., Tao F. Digital Twin Driven Green Material Optimal-Selection towards Sustainable Manufacturing // Procedia CIRP. 2019. Vol. 81, no. 1. P. 1290-1294.
- Noel Y., D'Arco P., Demichelis R., et al. On the use of symmetry in the ab initio quantum mechanical simulation of nanotubes and related materials // Journal of Computational Chemistry. 2010. Vol. 31, no. 4. P. 855-862.
- Civalleri B., D'Arco P., Orlando R., et al. Hartree-Fock geometry optimisation of periodic systems with the CRYSTAL code // Chemical Physics Letters. 2001. Vol. 348, no. 1. P. 131-138.
- Dovesi R., Pascale F., Civalleri B., et al. The CRYSTAL code, 1976-2020 and beyond, a long story // Journal of Chemical Physics. 2020. Vol. 152, no. 1. P. 204111.
- Бандура A., Эварестов Р. Неэмпирические расчеты кристаллов в атомном базисе с использованием интернет-сайтов и параллельных вычислений. Санкт-Петербург: Издательство Санкт-Петербургского университета, 2004. 228 с.
- Pascale F., Zicovich-Wilson C., Gejo F.L., et al. The calculation of vibrational frequencies of crystalline compounds and its implementation in the CRYSTAL code // Chemical Physics Letters. 2004. Vol. 25, no. 6. P. 888-897.
- Lohse M.S., Bein T. Covalent Organic Frameworks: Structures, Synthesis, and Applications // Advanced Functional Materials. 2018. Vol. 28, no. 33. P. 1705553.
- Биленко Р., Долганина Н., Иванова Е., Рекачинский А. Высокопроизводительные вы числительные ресурсы Южно-Уральского государственного университета // Вестник ЮУрГУ. Серия: Вычислительная математика и информатика. 2022. Т. 11, № 1. C. 15-30.
