Методика расчета электронной структуры ПВДФ и его карбонизованных производных

Бесплатный доступ
В работе проводится моделирование электронной структуры поливинилиденфторида (ПВДФ) в процессе карбонизации. В рассчитанной из первых принципов плотности состояний выделяется и исключается вклад концевых групп молекул, что позволяет повысить точность проводимых расчетов.
Короткий адрес: https://sciup.org/147158600
IDR: 147158600 | УДК: 544.163.2
Текст научной статьи Методика расчета электронной структуры ПВДФ и его карбонизованных производных
Поливинилиденфторид (ПВДФ) - полимер, который нашел широкое применение в промышленности благодаря своим свойствам (высокой механической прочности, эластичности, химической инертности, термостойкости и другим параметрам), а также перспективный материал для электроники и медицины.
Воздействия различной природы (рентгеновское излучение, бомбардировка заряженными частицами, химические воздействия) приводят к карбонизации поверхности полимера (отщеплению атомов фтора и водорода и образованию карбиновых фрагментов). Данный процесс представляет интерес с нескольких точек зрения - как деградация устойчивого к различного рода воздействиям полимера и как метод синтеза карбиновых структур [1, 2]. Исследование этого процесса позволит осуществлять контролируемое воздействие на полимер и получать материалы определенной структуры и свойств.
Экспериментальные работы не позволяют получить детальное представление о ходе карбонизации, поскольку фиксируют лишь определенные физико-химические параметры. Проведение компьютерного моделирования позволяет глубже рассмотреть течение процесса карбонизации и определить для него оптимальные условия.
Расчет электронной структуры проводится из первых принципов, поэтому необходимо учитывать ресурсозатратность этой процедуры, а именно достаточно высокие требования к производительности компьютера. Это накладывает ограничения на размеры систем для расчета. Другим ограничением является необходимость использования граничных условий, которыми в данном случае являются метиловые группы на концах молекулы.
Таким образом, задача сводится к расчету электронной структуры ограниченной молекулы с граничными условиями, что отличается от экспериментальных условий. Для того чтобы минимизировать это отличие, необходимо исключить из результатов расчета влияние концевых групп. Для этого из спектров, полученных по результатам оптимизации геометрии молекул, выделялись и исключались линии, соответствующие метиловым группам. Квантово-химические расчеты осуществлялись с использованием пакета программ GAMESS [3] на вычислительном кластере МВС-100 кафедры общей и теоретической физики Южно-Уральского государственного университета. Расчеты проводились методом функционала плотности (DFT) с использованием функционала B3LYP в базисе 6-31G. Был проведен расчет электронной структуры молекул ПВДФ различной степени карбонизации, а также межцепочечных сшивок между молекулами, которые, вероятно, образуются в ходе данного процесса.
На рис. 1 представлена плотность состояний остовного уровня углерода молекулы полиина, состоящей из 12 атомов углерода, из которых 2 принадлежат метиловым группам. Сплошной линией на графиках обозначена плотность состояний, из которой вычтен вклад СНз-групп, пунктирной - спектр, из которого влияние СНз-групп не вычиталось. На рис. 2 представлена плотность состояний остовного уровня углерода сшивки (состоящей из 14 атомов углерода, из которых 4 принадлежат метиловым группам). Из сравнения спектров видно, что метиловые группы дают ощутимый вклад в плотность состояний, пропорциональный отношению количества данных групп к общему количеству атомов углерода в молекуле.
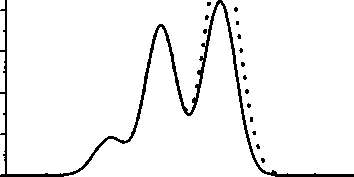
Даже при увеличении размера молекулы до 24 атомов углерода (из которых 4 атома принадлежат метиловым группам) вклад концевых групп все еще остается значительным (рис. 3). Дальнейшее увеличение размера молекулы приведет к снижению вклада СНз-групп, но при этом несоизмеримо увеличится и время расчета (для первопринципных расчетов зависимость времени
Химия
расчета от числа частиц возрастает от № до N4 в зависимости от конфигурации системы, где N -число атомов).
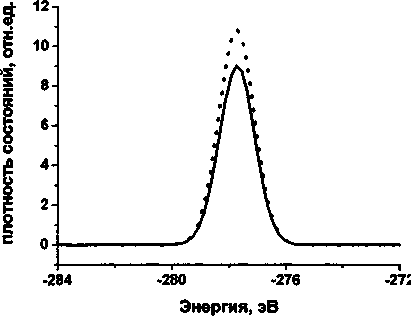
Рис. 1. Cis-спектр молекулы полиина из 12 атомов углерода
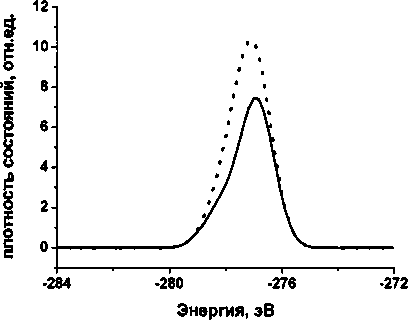
Рис. 2. Cis-спектр молекулы сшивки из 14 атомов углерода
12п
^284 ' -280 ' -276 ' ^272
Энергия, эВ
Рис. 3. Cis-спектр молекулы сшивки из 24 атомов углерода
Таким образом, при расчете электронной структуры молекул размерами до нескольких десятков атомов необходимо исключать влияние граничных условий на результаты вычислений. Это становится особенно важным при моделировании систем из небольшого числа частиц (порядка десятка атомов). При расчете электронной структуры данный подход позволяет получать достоверные результаты, не увеличивая при этом время расчетов.
Список литературы Методика расчета электронной структуры ПВДФ и его карбонизованных производных
- Pesin L.A. In situ observation of the modification of carbon hybridization in poly(vinilydene fluoride) during XPS/XAES measurements/L.A. Pesin et al.//Chemical Physics Letters. 2003. Vol. 372. P. 825-830.
- Okudaira K.K. Photodegradation of poly(tetrafluoroethylene) and poly(vinylidene fluoride) thin films by inner shell excitation/K.K. Okudaira et al.//Surface Review and Letters. 2002. Vol. 9, N 1. P. 335-340.
- Shmidt M.W. General atomic and molecular electronic structure system/M.W. Shmidt et al.//J. Comp. Chem. 1993. Vol. 14, N 11. P. 1347-1363.
