Моделирование из первых принципов сегрегации атомов железа на поверхности (111) никеля
 никеля")
Автор: Вяткин Герман Платонович, Морозов Сергей Иванович
Рубрика: Физика
Статья в выпуске: 2 т.7, 2015 года.
Бесплатный доступ
Работа посвящена компьютерному моделированию поверхности сплавов на основе Ni первопринципными методами. Получены модели поверхности сплавов Ni(111), рассчитаны их физические свойства: релаксация, поверхностная энергия, работа выхода. Исследовано влияние расположения атомов Fe на эти характеристики.
Моделирование, первопринципные методы, поверхность, никель, железо, сегрегация, релаксация
Короткий адрес: https://sciup.org/147158855
IDR: 147158855 | УДК: 669.24.056.9:004+539.211
Ab initio modeling of segregation of iron atoms on the (111) nickel surface
The paper is devoted to computer modeling of surface alloys Ni-based ab initio methods. Obtained surface model alloys Ni (111), calculated their physical properties: relaxation, surface energy, work function. Effect of location of the Fe atoms on these characteristics is studied.
Текст научной статьи Моделирование из первых принципов сегрегации атомов железа на поверхности (111) никеля
Поиск новых материалов, обладающих заданными свойствами, является одной из основных задач современной физической химии и материаловедения. При открытии перспективных направлений исследований количество научных работ, связанных с ними, как теоретических, так и экспериментальных резко возрастает. Подобное явление наблюдается в последнее десятилетие и с углеродными нанотрубками, и с нановолокнами. Активному использованию их уникальных свойств в промышленности, однако, мешает отсутствие доступной и дешевой технологии управляемого роста (синтеза) этих объектов. Для ее создания активно и продуктивно используются различные металлические катализаторы. В экспериментальной работе [1] отмечается, что как получаемое количество нанотрубок, так и характер их роста существенно зависит от используемой поверхности-катализатора Ni, Fe, Co. Для объяснения этого явления уже предложено несколько моделей (механизмов) роста углеродных нанотрубок (механизм карбидного цикла, модель «твердая фаза–жидкость–твердая фаза»). Их ключевым моментом является характер взаимодействия атомов углерода с поверхностными атомами. К сожалению, применение экспериментальных методов для изучения такого взаимодействия затруднено, но использование современных методов компьютерного моделирования из первых принципов позволяет исследовать его подробно и надежно. Согласно [2], в настоящее время число теоретических работ по изучению адсорбции атомов углерода на поверхности Ni и Fe невелико, при этом иногда они имеют противоречивый характер [3].
В настоящей работе проведены расчеты энергетики и структур моделей исследуемых поверхностных слоев сплавов Ni 20– x Fe x , которые в дальнейшем предполагается применить для изучения механизма взаимодействия атомов углерода, водорода и фрагментов молекул метана с поверхностью этих катализаторов.
Метод
Вычисления проводились с использованием лицензионной программы ab-initio расчета полной энергии и молекулярной динамики VASP (Vienna ab initio simulation program), разработанной факультетом физики Университета Вены [4, 5].
Расчет электронной структуры атомов выполнялся в рамках теории функционала электронной плотности (DFT) с использованием базиса плоских волн и формализма PAW, потенциалов обменно-коррекционного функционала PBE (Пердью–Бурке–Эрнцерхофа). Энергия обрезания базиса плоских волн была выбрана равной 500 eV.
Для исследования в качестве модели использовалась суперячейка (рис. 1), размерами 2 х 2 х 5 (5 слоев по 4 атома в слое). Атомы первого и второго слоя для корректного воспроизведения электронной структуры объема, были зафиксированы в равновесных «идеальных» позициях объемной решетки, атомы других «верхних» слоев могли релаксировать. Толщина вакуумного слоя составляла 10 Å, что исключало взаимодействие поверхностей из-за периодических граничных условий. Интегрирование в зоне Бриллюэна выполнялось по методу Монкхорста–Пака для K-сетки 9 х 9 х 1.
Релаксация получаемых структур проводилась методом сопряженных градиентов до достижения критериев сходимости по полной энергии системы менее 10–5 эВ и до величины остаточной силы, действующей на ион, менее 10–3 эВ/Å. Использованные значения параметров расчетной схемы были достаточны для обеспечения надежности результатов.
Для обозначения положения атомов Fe используется нумерация атомов Ni в ячейке. Для ви- зуализации моделей использовалась программа VESTA [6].

а) вид сверху; б) вид сбоку
Рис. 1. Модель исследуемой поверхности Ni (111)
Результаты и их обсуждение
В качестве начальной модели для исследования была принята оптимизированная модель Ni(111), в которой последовательно один атом никеля в позициях с 9 по 20 замещался атомом железа. Полученные значения полной энергии структур и их отличие от минимально возможной энергии представлены в табл. 1.
Таблица 1
Зависимость полной энергии структур Ni 11 Fe от расположения атома Fe
|
Положение атомов Fe |
Полная энергия, эВ |
Приращение энергии, эВ |
|
13 |
–107,35 |
0,00 |
|
14 |
–107,35 |
0,00 |
|
15 |
–107,35 |
0,00 |
|
16 |
–107,35 |
0,00 |
|
9 |
–107,27 |
0,08 |
|
10 |
–107,27 |
0,08 |
|
11 |
–107,27 |
0,08 |
|
12 |
–107,27 |
0,08 |
|
17 |
–107,16 |
0,19 |
|
18 |
–107,16 |
0,19 |
|
19 |
–107,16 |
0,19 |
|
20 |
–107,16 |
0,19 |
Наиболее энергетически выгодным является расположение атомов в четвертом слое в эквивалентных по энергиям положениях 13, 14, 15, 16. Далее менее приоритетными являются места в третьем слое (позиции 9–12). Нахождение атома замещения в пятом поверхностном слое (позиции 17–20) вызывает наибольшее увеличение энергии структуры. В качестве базовой для дальнейших исследований была выбрана структура I с минимальной энергией и с расположением атома железа в положении 13 (рис. 2, а ).
Физика
Затем в структуре I последовательно замещался атом никеля атомом железа в положениях 9, 10, 11, 12, 14, 15, 16, 17, 18, 19, 20 и выполнялся расчет полной энергии полученных структур. Результаты расчета, упорядоченные по возрастанию энергий, представлены в табл. 2.
Наименьшая энергия для изучаемой структуры была получена при нахождении атома Fe в третьем слое (положение 11), на максимально возможном удалении от атома 13. Эта модель – структура II и будет использована далее. Равновероятными (эквивалентными) в плане энергии являются позиции в третьем слое – 9, 10, 12 и в верхнем – 18. Наибольшую энергию имеет рассматриваемая структура при замещении атомов пятого слоя (позиции 17, 19, 20).
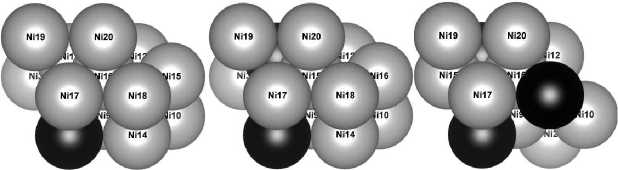
Вид сверху
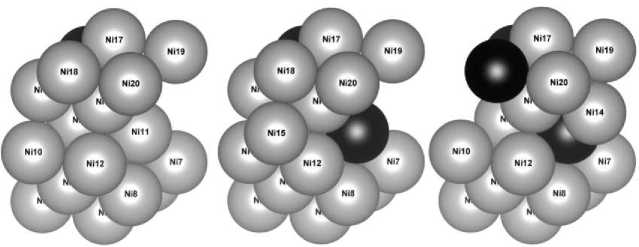
Вид сбоку
а) x = 1; б) x = 2; в) x = 3.
Рис. 2. Наиболее энергетически выгодные модели поверхностного слоя Ni 12– x Fe x атомы Ni показаны серым цветом, атомы Fe черным
Таблица 2
Зависимость полной энергии структур Ni 10 Fe 2 от расположения второго атома Fe (первый атом Fe зафиксирован в положении 13)
|
Положение атомов Fe |
Полная энергия, эВ |
Приращение энергии, эВ |
|
11 |
–110,48 |
0,00 |
|
18 |
–110,32 |
0,16 |
|
9 |
–110,31 |
0,17 |
|
10 |
–110,31 |
0,17 |
|
12 |
–110,31 |
0,17 |
|
14 |
–110,29 |
0,19 |
|
15 |
–110,29 |
0,19 |
|
16 |
–110,29 |
0,19 |
|
17 |
–110,14 |
0,34 |
|
19 |
–110,14 |
0,34 |
|
20 |
–110,14 |
0,34 |
Для подтверждения нахождения структуры с минимальной энергией были проведены дополнительные расчеты (табл. 3), в которых один атом железа в позиции 11 фиксировался, а второй мог занимать произвольные равновесные позиции атомов в приповерхностных слоях. Как следует из результатов, структура II (атомы Fe в положениях 11 и 13) действительно является оптимальной и в этом случае.
Таблица 3
Зависимость полной энергии структур Ni 10 Fe 2 от расположения второго атома Fe (первый атом Fe зафиксирован в положении 11)
|
Положение атомов Fe |
Полная энергия, эВ |
Приращение энергии, эВ |
|
13 |
–110,48 |
0,00 |
|
14 |
–110,31 |
0,17 |
|
15 |
–110,31 |
0,17 |
|
16 |
–110,31 |
0,17 |
|
17 |
–110,21 |
0,27 |
|
19 |
–110,21 |
0,27 |
|
20 |
–110,21 |
0,27 |
|
18 |
–110,19 |
0,29 |
|
9 |
–110,11 |
0,37 |
|
10 |
–110,11 |
0,37 |
|
12 |
–110,11 |
0,37 |
Аналогичные манипуляции по размещению третьего атома Fe были произведены со структурой II. Результаты расчета полной энергии представлены в табл. 4.
Исходя из представленных расчетов, наиболее энергетически выгодной является структура III (рис. 2, в ), в которой третий атом размещен в положении 18 (пятый слой), на максимально возможном в данной модели удалении от атома соседнего слоя. Наибольшей энергией структура обладает при замещении атомов никеля в третьем слое в положениях 9, 10, 12.
Таблица 4
Зависимость полной энергии структур Ni 9 Fe 3 от расположения атома Fe
|
Положение атомов Fe |
Полная энергия, эВ |
Приращение энергии, эВ |
|
18 |
–113,42 |
0,00 |
|
14 |
–113,30 |
0,12 |
|
15 |
–113,30 |
0,12 |
|
16 |
–113,30 |
0,12 |
|
17 |
–113,25 |
0,17 |
|
19 |
–113,25 |
0,17 |
|
20 |
–113,25 |
0,17 |
|
9 |
–113,20 |
0,22 |
|
10 |
–113,20 |
0,22 |
|
12 |
–113,20 |
0,22 |
По полученным моделям поверхностного слоя I, II и III были вычислены следующие физические характеристики (табл. 5): поверхностная энергия, работа выхода электронов, относительное изменение расстояния между слоями поверхности по отношению с расстояниями в объеме (релаксация поверхностного слоя), энергия Ферми.
Таблица 5
Характеристики моделей поверхности
|
Состав |
Релаксация поверхностного слоя, % |
Толщина слоев, Å |
Поверхностная энергия, эВ |
Работа выхода электрона, эВ |
Энергия Ферми E F , эВ |
||
|
δ 23 |
δ 34 |
δ 45 |
|||||
|
Ni(111) |
–0,32 |
–0,22 |
–1,45 |
8,09 |
0,64 |
4,69 |
3,02 |
|
Ni 11 Fe |
0,03 |
–0,73 |
–0,57 |
8,10 |
0,58 |
4,65 |
2,97 |
|
Ni 10 Fe 2 |
0,24 |
0,14 |
–1,08 |
8,12 |
0,51 |
4,63 |
2,90 |
|
Ni 9 Fe 3 |
0,08 |
0,86 |
–0,90 |
8,13 |
0,47 |
4,70 |
2,83 |
Физика
Замена атома никеля атомом железа приводит к изменению расстояний между слоями. Так, расстояние между вторым и третьим слоем в структурах I, II, III, больше объемного расстояния, в то время как в чистом металле оно меньше. Сжатие для всех структур наблюдается только между слоями 4 и 5. В целом, толщина приповерхностного слоя с добавлением атомов железа увеличивается с 8,09 Å до 8,13 Å .
В полученных моделях наблюдается уменьшение поверхностной энергии от 0,64 эВ (чистая поверхность Ni(111)) до 0,47 эВ (структура III). Энергия Ферми также убывает. Средний магнитный момент атомов Ni и Fe не изменяется. Работа выхода электрона сначала убывает от 4,69 эВ (чистая поверхность), достигая минимального значения 4,63 эВ (структура II), затем резко возрастает до 4,70 эВ (структура III).
Для структуры I, II и III были рассчитаны плотности электронных состояний (рис. 3). Наличие атомов железа проводит к их перераспределению. Так последовательное увеличение количества атомов Fe уменьшает плотность состояний для энергий –1,8; –0,8; и 6,9 эВ для конфигурации «спин вверх» и –1,4; 0,1; 0,8; 7,0 и 7,7 эВ для конфигурации «спин вниз».
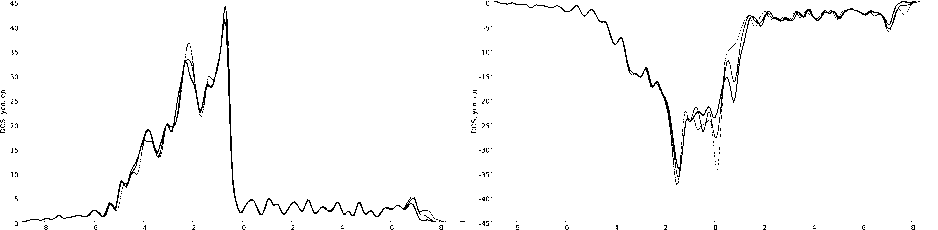
а) спин «вверх»; б) спин «вниз».
Рис. 3. Плотность электронных состояний исследуемых моделей
Толщина линии показывает количество атомов Fe:
самая тонкая линия соответствует «чистой» поверхности Ni (111), самая толстая – структуре с тремя атомами Fe
Заключение
Выполненные расчеты в рамках теории функционала электронной плотности позволили получить модели поверхностных слоев сплавов Ni (111) с различным содержанием атомов Fe, которые в дальнейшем предполагается использовать для расчета энергий и геометрических параметров при адсорбции атомов и молекул. Выявлено влияние содержания атомов Fe в исследуемых моделях на изменение физических характеристик поверхностного слоя. Их увеличение приводит к снижению поверхностной энергии, к снижению работы выхода электрона для структур I и II и ее росту для структуры III, релаксации расстояний между атомными слоями, вызывает изменение электронной структуры.
Наиболее энергетически выгодной для адсорбции частиц является структура III, содержащая три атома Fe, размещенных на максимальном удалении друг от друга в «верхних» слоях.
Авторы благодарят директора лаборатории суперкомпьютерного моделирования ЮУрГУ П.С. Костенецкого, директора суперкомпьютерного центра ЮУрГУ К.В. Бородулина за всестороннюю техническую поддержку при проведении исследований.
Список литературы Моделирование из первых принципов сегрегации атомов железа на поверхности (111) никеля
- Исследование микроструктуры углеродного наноматериала, полученного на железо-никелевом катализаторе/А.А. Новикова, Т.Ю. Киселева, Б.П. Тарасов, В.Е. Мурадян//Поверхность. Рентгеновские, синхротронные и нейтронные исследования. -2004. -№ 3. -С. 70-73.
- Мутигуллин, И.В. Влияние степени покрытия углеродом на возможность формирования твердого раствора внедрения в подповерхностном слое железа (001) и (111)/И.В. Мутигуллин, Д.И. Бажанов, А.С. Илюшин//Физика твердого тела. -2011. -Том 53. -Вып. 3. -С. 558-563.
- Mueller, J.E. Structures, Energetics and Reaction Barriers for CHx Bound to the Nickel (111) Surface/J.E. Mueller, A.C.T. van Duin, W.A. Goddard, III//J. Phys. Chem. C. -2009. -Vol. 113, № 47. -P. 20290-20306.
- Kresse, G. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set/G. Kresse, J. Furthmüller//Phys. Rev. B. -1996. -Vol. 54. -Issue 16. -P. 11169.
- Kresse, G. From ultrasoft pseudopotentials to the projector augmented-wave method/G. Kresse, D. Joubert//Phys. Rev. B -1999. -Vol. 59. -Issue 3. -P. 1758.
- Momma K. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data/K. Momma, F. Izumi//Journal of Applied Crystallography. -2011. -Vol. 44. -Issue 6. -P. 1272-1276.
