Molecular heterogeneity of classical and Duarte galactosemia

Author: Aliyeva Kamila, Hajiyeva Nilufar
Journal: Бюллетень науки и практики @bulletennauki
Section: Медицинские науки
Article in issue: 8 т.8, 2022.
Free access
For the first time we have conducted molecular-genetic analysis of GALT gene of galactosemia inherited disease in the newborn blood which found at the result of screening conducted maternity houses of Baku city of Azerbaijan Republic. For Azerbaijan people for the first-time identification of substitution of adenine nucleotide with guanine nucleotide in GALT gene position 563 was conducted. It leaded to substitution of glycineamino acid with arginine amino acid in position 188 of mutation protein. Galactosemia is an alteration that alters the way the body processes galactose. It may be due to a deficiency of any of the three enzymes of the galactose catabolic pathway; galactose-1- phosphate urydiltransferase (Gal-1-PUT), galactokinase or urydil diphosphate (UDP)- galactose-4-epimerase. Individuals having galactosemia are intolerant to lactose and galactose in milk. In the lack of proper diagnosis and treatment, this disorder can lead to enlarged liver, cirrhosis, renal failure, cataracts, brain damage and even death. Long term complications can be prevented with early diagnosis following birth and by eliminating lactose and galactose containing nutrients from daily diet.
Galactosemia, metabolic disorder, galactose-1-phospaturidyltransferase, polymerase-chain reaction, mutation
Short address: https://sciup.org/14124762
IDR: 14124762 | UDC: 612.6.05 | DOI: 10.33619/2414-2948/81/16
Молекулярная гетерогенность классической галактоземии и галактоземии Дуарте
Впервые проведен молекулярно-генетический анализ гена ГАЛТ наследственного заболевания галактоземии в крови новорожденных, обнаруженного по результатам скрининга родильных домов города Баку Азербайджанской Республики. Для азербайджанцев впервые была проведена идентификация замещения аденинового нуклеотида гуаниновым нуклеотидом в положении 563 гена GALT. Он приводил к замещению глицинаминовой кислоты аргининовой аминокислотой в положении 188 мутационного белка. Галактоземия изменяет способ обработки галактозы организмом. Это может быть связано с дефицитом любого из трех ферментов катаболического пути галактозы; галактоза-1- фосфатуридилтрансфераза (Гал-1-ПУТ), галактокиназа или уридилдифосфат (УДП) - галактоза-4-эпимераза. Люди с галактоземией непереносят лактозу и галактозу в молоке. При отсутствии правильной диагностики и лечения это расстройство может привести к увеличению печени, циррозу, почечной недостаточности, катаракте, повреждению мозга и даже смерти. Длительные осложнения можно предотвратить с помощью ранней диагностики после рождения и путем исключения лактозы и галактозосодержащих питательных веществ из ежедневной диеты.
Text of the scientific article Molecular heterogeneity of classical and Duarte galactosemia
Бюллетень науки и практики / Bulletin of Science and Practice Т. 8. №8. 2022
UDC 612.6.05
Mutations that severely impair enzyme activity result in the classic galactosemia phenotype. If a child with GALT eats galactose, undigested sugars build up in the blood rather than being used for energy. The GALT (galactose-1- phosphate uridylytransferase) gene, locatedon the short arm of chromosome 9(9p13), galactose-1- phosphate. A mutation of GALT gene knows as a the Duarte variant (Asn 314 Asp or N314D), reduces, but does not eliminate the activity of the enzyme. People with the Duarte variant usually have much milder sign and symptoms of galactosemia because the enzyme retains 5 to 20 percent of its normal activity. If GALT is left untreated, it can cause seizures, serious blood infections, liver damage, or even death. However, when the condition is identified early in life and proper treatment is begun immediately, children with GALT often can lead healthy lives. This hereditary condition is passed from parent to child as an autosomal recessive disease. This means that a child needs to inherit two copies of the defective gene (one from each parent) in order to have the disease. By galactose-1-phosphateurydiltransferase enzyme deficit, the metabolism of galactose disaccharide: galactose cleavage into two glucose molecules is damaged. Cataract, hepatomegaly following with liver cirrhosis is observed in patients with later development of physical and mental retardation (1,2,3,4). Genetics of galactosemia disease is heterogeneous. Genes participating in galactose metabolism are located in the chromosomes 1, 9 and 17. GeneGAL1 is located in p13 of the short shoulder in chromosome 9. Gene GALKis positioned in siteq23-q25 of the long shoulder of chromosome 17, and gene GALЕ is located in the short shoulder of the chromosome 1 in site p35-p36. Inheritance type for all three types of galactosemia gene is autosome-recessive. Frequency for homozygotes is 1:15000-20000, and for heterozygotes 1:270-300 newborns (5,8,9,10).
In Russian Federation the following mutations for galactosemia gene are revealedandi dentified: GALT substitution of adenine nucleotide with guanine nucleotide in position 563, leading to substitution of glycine amino acid with arginine amino acid in position 188; and substitution of guanine nucleotide with adenine nucleotide in position184, resulting in leucine amino acid substitution with methionine amino acid in position 62. In the USA, patients with mental retardation manifested these mutations both in homozygous and compound state (2,9,10). Molecular-genetic diagnostics of GALT gene of galactosemia inherited disease in Azerbaijan Republic people is not conducted till now. Aim of our study was DNA analysis of GALT gene founded at the result of screening conducted among newborns.
Materials and Methods
Material for our research was venous blood 2-ml sample from a newborn A.M. on anticoagulant EDTA. Molecular-genetic methods based on polymerase-chain reaction (PCR) and electrophoresis of genomic DNA, amplified DNA fragments and different fragments nucleotides were used for mutations’ identification. Genomic DNA was isolated form venous blood sample by means of readymade QIA amp genomic DNA and RNA kits, product of QIAGEN, Germany. Intactness and quantity of the isolated genomic DNA as well as of the amplificate (gene fragment) after PCR were identified with electrophoresis in 1.7% agarose gel by means of electrophoretic apparatus and power source (Power Pac Basic GelDocIMEZ) Imager, made byBioRad, USA. During electrophoresis, DNA
Ladder 100 bp was used as a marker for synthesized DNA fragments identification. PCR regime for gene GAL1 was 95оС-2 minutes, (95оС-30I, 60оС-30I, 76оС-2 minutes X 30 cycles), 72оС for 10 minutes and break at 4оС for 10 minutes. PCR was conducted in amplifier Professional Thermocycler manufactured by Biometra, Germany. Two primers (Forward и Reverse) were used to amplify each site of GALT (5 exons). For purification of DNA fragments after the first PCR step there was used a set of magnets: «A gen court AMPure XP PCR purification» and SPRIP late 96 Super Magnet Plate Purified DNA fragments were applied for next researches. The second PCR step was carried out in the following regime: 95оС for 2 minutes, (95оС-30I , 52оС-58оС– 30I, 78оС-2 minutes X 30 cycles), 72оС for 10 minutes and pause in the amplifierat 4оС for 10 minutes. Then a standard procedure was done in the GENOME Lab GeXPTM Sequencing apparatus to identify nucleotide sequences for each DNA fragment (6,8).
Results and discussion
Analysing of DNA molecule obtained from lymphocytes of newborn A.H.
1. It was defined the deletion of 4 nucleotide (c. 119–116 del GTCA) of promotor area of GALT gene of galactose-1-phosphaturidiltransphetase. Type of this genetic modification was determined as
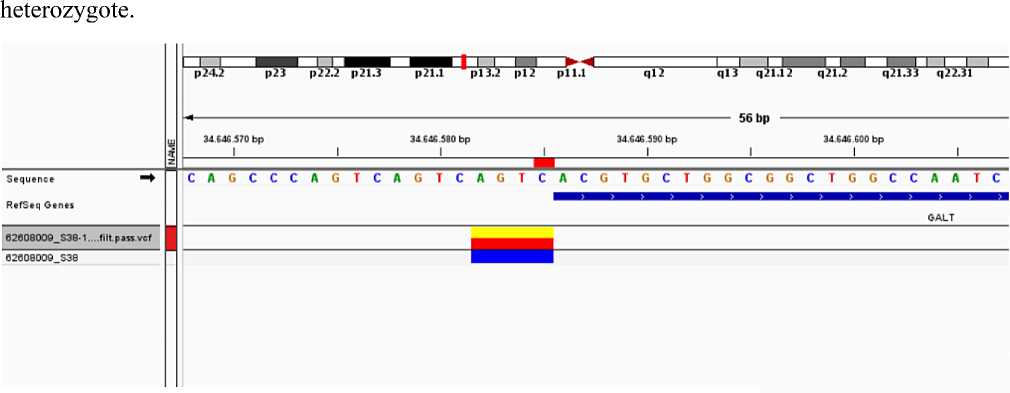
2. It was also defined single nucleotide polimorphism characterised replacing of cytosine with thymine at 625th codon (C > T) at 7th exon of GALT gene of the same pacient. Clinically, this genetical modification was characterised as genetical version which is not cause to changes of sinless and
3. Another mutation of pacient was replacing of adenine with guanine at 940-codon of 10th exon at the GALT gene (940 A>G). Together observing of -119-116 del GTCA deletion which found at the promotor area of GALT gene were given in world-wide references pathogenically. At 314th position of protein Aspartic amino-acid was replaced with aspargin amino-acid (c.940 A >G; p Asn 314 Asp).
It was founf that type of this genetic modification is homozygote.
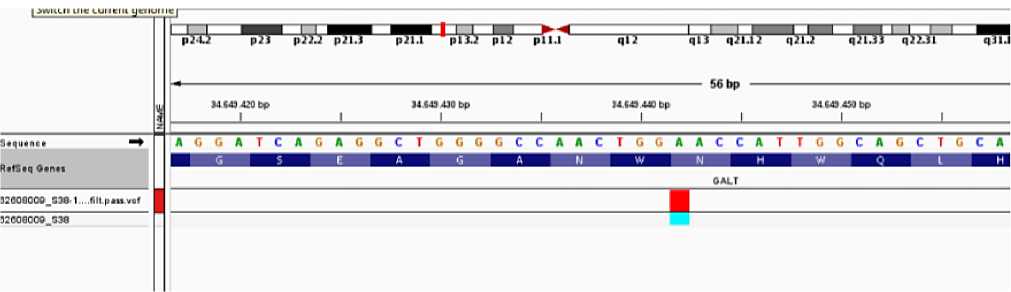
Observing of both forms at GALT gene- was accepted as a genetic reason of compaund galatosemia disease. These changes are pathological modifications approving galactosemia disease. Mutation also found at the 3th subsequent of 13th segment of the small side (p) of 9th chromosome.
-
1. During molecular-genetic analysis of GALT gene of the second newborn A.K. it was found deletion at the beginning area of the 1st exon of 9th chromosome of gene.

-
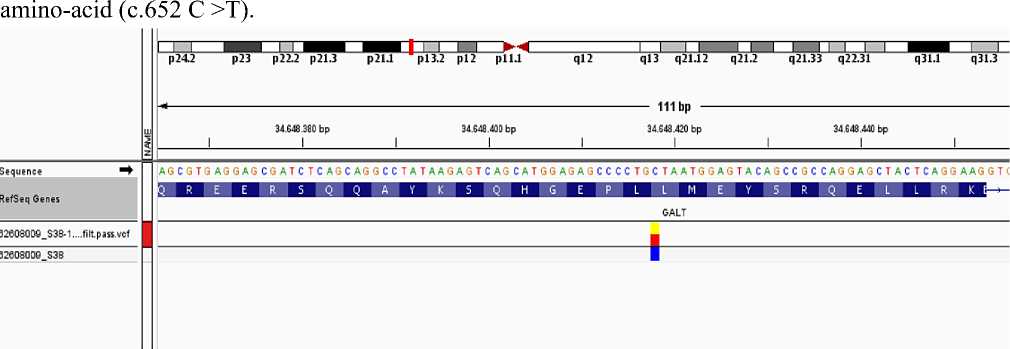
2. Single nucleotide polymorphism at the 7th exon of GALT gene, at c.652 codon of C >T protein p (Leu 218) part replacing of one nucleotide occured. Replacing of cytosine with thymine C >T at c.652 codon caused to mutation. Type of this genetic modification was determined as heterozygote. It is at the 3rd subsequent of (p)13 sequent of the smail side of 9th chromosome. It doesn’t effect to protein function clinically.
it genome]
-
' I I ^^■^^^^■~^^ | I I 14 H I I I I— I I- I I-.......^И—I I ■» ......
34.648 400 bp 34.648.410 bp 34.648.420 bp 34 648 .430 bp 34 648 440 bp
p24.2 p23 1122.2 p2L3 p21.1 pl3.2 pll.l ql2 ql3 q21.12 q21.2 q21.32 q22.1 q22.32 q31.1 q31.3 q33.1 q33.3 q34.12 q-
1 ^------------------------------------------------------------------------- 54 bp ------------------------------------------------------------------------
GGCCTATAAGAGTCAGCATGGAGAGCCCCTGCTAATGGAGTACAGCCGCCAGG
GALT
References Molecular heterogeneity of classical and Duarte galactosemia
- Bosch A. M. Classic galactosemia: dietary dilemmas // Journal of inherited metabolic disease. 2011. V. 34. №2. P. 257-260.
- Bennett M. J. Galactosemia diagnosis gets an upgrade // Clinical chemistry. 2010. V. 56. №5. P. 690-692.
- Shield, J. P. H., Wadsworth, E. J. K., MacDonald, A., Stephenson, A., Tyfield, L., Holton, J. B., & Marlow, N. The relationship of genotype to cognitive outcome in galactosaemia // Archives of disease in childhood. 2000. V. 83. №3. P. 248-250.
- Galactosemia I type. Classic galactosemia. Diagnosis of galactosemia. Clinics of galactosemia. Medician house.
- Report of scientific group of World Health Organization // Fight with hereditary diseases. Jeneva. 1997. P. 133.
- Gathof B. S., Sommer M., Podskarbi T., Reichardt J., Braun A., Gresser U., Shin Y. S. Characterization of two stop codon mutations in the galactose-1-phosphate uridyltransferase gene of three male galactosemic patients with severe clinical manifestation // Human genetics. 1995. V. 96. №6. P. 721-725.
- Application in formation. Purification of GENOME LABTM GeXPSequencing Productions using SPRIClean SEQRMagnetic Beards. CEQ 2000, CEQ 000XL, CEQ 8000, CEQ 8800 &GeXP Instruments BECKMAN COULTER. Application Team Europe.
- Beutler E. G6PD deficiency // Blood. 1994. V. 84. №11. P. 3613-3636.
- Gu W., Zhang F., Lupski J. R. Mechanisms for human genomic rearrangements // Pathogenetics. 2008. V. 1. №1. P. 1-17.
- Herman R., Kunisaki S., Molitor M., Gadepalli S., Hirschl R., Geiger J. The use of peritoneal venous shunting for intractable neonatal ascites: a short case series // Journal of pediatric surgery. 2011. V. 46. №8. P. 1651-1654.
