Молекулярно-генетический анализ генов GJB2, SLC26A4 и SLC26A5 у больных с наследственными нарушениями слуха из Республики Башкортостан

Автор: Лобов Семен Леонидович, Джемилева Лиля Усеиновна, Хуснутдинова Эльза Камилевна
Журнал: Известия Самарского научного центра Российской академии наук @izvestiya-ssc
Рубрика: Генетика
Статья в выпуске: 5-3 т.13, 2011 года.
Бесплатный доступ
Рассмотрен анализ генов GJB2, SLC26A4 и SLC26A5 у больных с наследственными нарушениями слуха, проведён сравнительный анализ распространения данных мутаций в разных странах мира.
Глухота, внутреннее ухо, гены
Короткий адрес: https://sciup.org/148205563
IDR: 148205563 | УДК: 575.176,
Molecular genetic analysis of GJB2, SLC26A4 and SLC26A5 genes in patients with hereditary hearing loss from Bashkortostan
In this article the analysis of genes GJB2, SLC26A4 and SLC26A5 in patients with hereditary hearing impairment, a comparative analysis of data distribution of mutations in different countries.
Текст научной статьи Молекулярно-генетический анализ генов GJB2, SLC26A4 и SLC26A5 у больных с наследственными нарушениями слуха из Республики Башкортостан
Человеческое ухо способно воспринимать звуки от 20-20000 Гц и может различать разницу всего лишь на 0,2-0,5% [4, 6]. Такая чувствительность и точность, наблюдающаяся и у других млекопитающих, происходит от сложного взаимодействия кохлеарных волосковых клеток и текториальной мембраны в пределах кортиева органа. При нарушении этого взаимодействия наступает глухо-та/тугоухость.
Последние исследования показали, что более 75% детской глухоты генетического происхождения [1]. Около четверти генетических форм син-дромальные (глухота является частью генетического синдрома). Наиболее частая форма наследственной глухоты – несиндромальная сенсоневральная тугоухость/глухота (НСНТ). В разных странах мира из 1000 новорожденных 1 ребенок рождается глухим, и глухота в основном прелингвальная [22]. По статистическим данным ВОЗ, в мире насчитывается около 300 млн человек, страдающих нарушением слуха различной этиологии (III-IV степени тугоухости). В РФ этот показатель превышает 13 млн человек, из которых более 1 млн. – это дети в возрасте до 18 лет.
На сегодняшний день определены более 80 локусов для несиндромальных форм потери слуха. Наиболее значимыми среди всех идентифицированных генов, вовлеченных в функционирование системы звуковосприятия, являются гены белков-коннексинов 26 ( GJB2 ), 30 ( GJB6 ) и 31 ( GJB3 ), мутации в которых, по данным разных авторов, являются причинами 50-80% случаев развития несин-дромальных и некоторых синдромальных форм глухоты/тугоухости [5, 27] и около 10-12% всех случаев потери слуха обусловлены повреждением генов белков SLC26A4 (пендрина) и SLC26A5 (престина) [11, 26].
Ген SLC26A4 (7q31) состоит из 21 экзона и кодирует белок пендрин, который является переносчиком I-, Cl- и CO 3 2- ионов [9,23]. Повреждение пендри-на приводит к развитию синдрома Пендреда (1/7500, ответственен за 5-10% всех врожденных
форм тугоухости) либо к неполному развитию костного и перепончатого лабиринтов улитки – «дисплазия Мондини» (расширение вестибулярного водопровода) [10]. На сегодняшний день известно около 100 мутаций в гене SLC26A4. Спектр и частота отдельных мутаций гена обладают этнической специфичностью. p.Thr416Pro и c.1001+1G>A – две наиболее частых мутации у пациентов из Северной Европы. У больных из Китая мутация c.919-2A>G выявляется на 57,63% хромосом пациентов с синдромом Пендреда и дисплазией Мондини. У больных из Японии преобладающей являются мутация p.His723Arg, которая обнаруживаются у 53% пациентов с синдромом Пендреда и дисплазией Мондини. Мутации p.His723Arg и c.919-2A>G также являются преобладающими у больных из Кореи, которые обнаруживаются у 45,5% пациентов с синдромом Пендреда и дисплазией Мондини [25].
Ген SLC26A5 (7q22.1), состоящий из 21 экзона, имеющий 4 сплайсинговые изоформы, высокогомологичен членам семейства сульфатных и анионных переносчиков, кодирует белок престин, который, приблизительно, на 40% гомологичен пендри-ну, но имеет несколько другие свойства [38]. Считается, что престин является двигательным белком наружных волосковых клеток. Мажорной мутацией в гене SLC26A5 для большинства пациентов из Европы является мутация g.-53-2A>G, выявляемая у 3% больных с нарушениями слуха.
Несмотря на актуальную медико-социальную значимость проблемы, данные о спектре мутаций в генах GJB2, SLC26A4 и SLC26A5 у больных с потерей слуха из различных этнических групп Республики Башкортостан (РБ) до сих пор не были получены. Цель нашего исследования – изучение спектра мутаций в генах GJB2, SLC26A4 и SLC26A5 у индивидов с нормальным слухом и у пациентов с тугоухостью/глухотой из различных этнических групп, проживающих на территории РБ.
МАТЕРИАЛ И МЕТОДЫ
Материалом для исследования послужили 246 образцов ДНК больных с диагнозом «наследственная несиндромальная тугоухость/глухота». По сте- пени потери слуха у пробандов семьи распределились следующим образом: I степень тугоухости была зарегистрирована в 4 семьях, II степень тугоухости – в 17 семьях, III степень тугоухости – в 31 семье, IV степень тугоухости – в 62 семьях, и глухота - в 132 семьях. Этнический состав обследованных молекулярно-генетическими методами семей был следующим: русские – 98 семей, татары – 58 семей, башкиры – 37 семей, мари – 5 семей, украинцы – 3 семьи, армяне – 3 семьи, метисы – 42 семьи.
Диагноз глухоты или тугоухости устанавливался в соответствии с современными диагностическими критериями, предлагаемыми Институтом глухоты и коммуникативных расстройств (г. Омаха, США) и рекомендованных в 2003 г. Европейской рабочей группой по наследственным нарушениям слуха GENDEAF [20,31,32]. Наследственный характер глухоты в семьях устанавливали на основании генеалогических данных и ретроспективного анализа анамнеза больных с целью исключения возможного влияния факторов внешней среды (инфекции, травмы слухового аппарата) во время пренатального и постнатального развития.
Образцы крови были взяты с информированного письменного согласия пациентов и их родителей. ДНК выделяли из 10 мл периферической крови методом фенол-хлороформной экстракции. Поиск мутаций в генах GJB2, SLC26A4 и SLC26A5 осуществляли с помощью ПЦР с последующим ПДРФ-анализом и анализа конформационного полимор-
272 П.Н.
112 П.Н.
91 П.Н.
69 л.н.
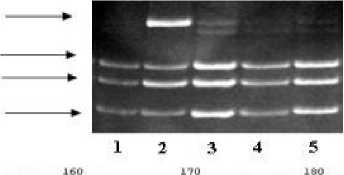
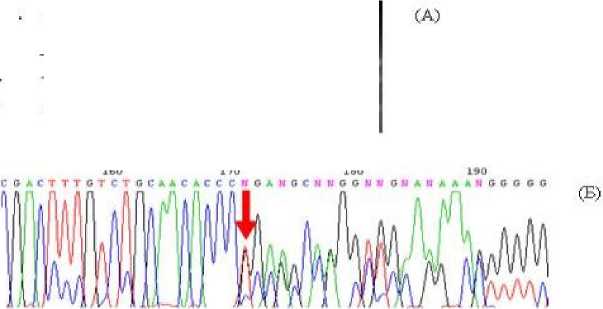
Рис. 1. Идентификация мутации с.167delT в гетерозиготном состоянии в гене GJB2 с помощью ПДРФ-анализа (Pst1) (А) и ресеквенирования 2 экзона гена GJB2 (Б) . Дорожки: 1, 3-5: образцы с генотипом norm/norm., 2 – образец с генотипом с.167delT/N.
Мутация с.167delT в гене GJB2 была идентифицирована у пробандов из 8 семей с НСНТ. В 4 семьях (2 метисных тат/рус, 1 русской и одной татарской) с.167delT выявлена в гетерозиготном состоянии, в 3 семьях (2 русских и 1 татарской) данная делеция была выявлена в компаунд-гетерозиготном состоянии с мутацией с.35delG. И в 1 татарской семье (Б) у пробанда с глухотой мутация с.167delT в гене GJB2 выявлена в сочетании с мутацией q.919-2A>G, расположенной в 7 интроне в сайте сплайсинга 8 экзона в гене SLC26A4 .
физма однонитевой ДНК (SSCP-анализ). Окрашивание конформеров проводили 0,1%-ным раствором AgNO3. Определение последовательности ДНК в образцах с аномальной электрофоретической подвижностью при SSCP-анализе проводили мощью автоматического секвенирования PRISM 310, ABI3100, Applied Biosystoms).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Мутация 167delT в гене GJB2 впервые с по-(АВ1
была
идентифицирована в 1998 г. у больных наследственной формой потери слуха в нескольких семьях евреев Ашкенази [21]. Далее различными исследователями были показаны достаточно высокие частоты этой мутации и в других популяциях Европы, Средиземноморья, Ближнего Востока, Северной Америки и Евразии [3, 21]. Всё вышеперечисленное послужило основанием для поиска с.167delT в выборке больных из РБ. При делеции с.167delT происходит потеря тимина в 167 положении, что приводит к сдвигу рамки считывания и образованию преждевременного стоп-кодона. Для детекции мутации с.167delT был подобран метод идентификации с помощью ПЦР с последующим ПДРФ– анализом. При иcпользовании данного метода теряется один из двух сайтов рестрикции для эндонуклеазы Pst1 (рис. 1), что позволяет выявлять гомозиготных и гетерозиготных носителей этой де-леции. Наличие мутации в образцах подтверждалось последующим ресеквенированием образцов.
Родословная пробанда представлена на рис. 2. У пробанда данной семьи четверо детей, которые также глухие, родители пробанда слышащие. К сожалению, образцы ДНК детей, жены и родителей пробанда оказались недоступны для исследования (родители пробанда, а также его жена и дети отказались проходить медико-генетическое консультирование по религиозным причинам).
Подобное сочетание мутаций g.-3179G>A и с.35delG в гене GJB2 и g.2-2A>G в гене SLC26A4 обнаружено в 2 эстонских семьях с нарушением слуха [34]. Также в литературе описаны случаи со- четания мутаций в гене GJB2 с протяженными де-лециями в гене GJB6 - del(GJB6-D13S1830) и del(GJB6-D13S1854): с.167 delT/del(GJB6-D13S1830) и с.35delG/del(GJB6-D13S1830) у итальянцев [28].
Частота мутации с.167delT в выборке больных из РБ составляет 2%. Во всех обследованных семьях наблюдается аутосомно-рецессивный тип наследования потери слуха, что согласуется с данными по частоте и этнической специфичности с.167delT [3, 21].
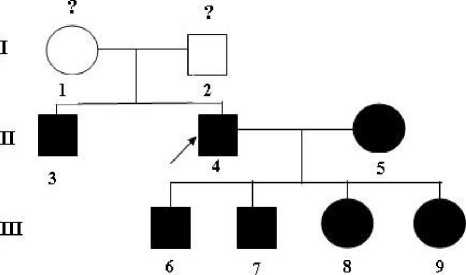
Рис. 2. Фрагмент родословной семьи Б. II-3 пробанд -глухой, генотип с.167delT (ген GJB2)/ c.919-2A>G (ген SLC26A4 ). Родители пробанда (мать I-1 и отец I-2) имеют нормальный слух.
Впервые трансверсия в 101 положении гена GJB2 описана в 1997 г. как доминантная мутация, вызывающая потерю слуха [16]. Замена тимина на цитозин в 101 положении приводит к замене полярной аминокислоты метионин на гетероциклическую – триптофан в 34 положении трансмембранного домена – Тм-1 белка коннексина 26. Далее в 1998 году c.101T>C была обнаружена в гетерозиготном состоянии в выборке 192 индивидов с нормальным слухом [15]. Однако несколькими иссле- дователями при изучении экспрессии коннексина 26 с заменой метионина на триптофан в 34 положении был подтвержден доминантно негативный эффект мутации c.101T>C in vitro [2, 8, 12, 36]. Таким образом, функциональная значимость данной замены на сегодняшний день спорна: ряд исследователей считает, что c.101T>C полиморфный вариант [17,33], а другие придерживаются мнения, что c.101T>C - мутация с аутосомно-рецессивным типом наследования [2, 8, 18].
Мутация c.101T>C выявлена у пациентов с НСНТ из Австралии (около 1%) [7], Австрии (0,98%) [14], Финляндии (3,8%) [19], Венгрии (0,28%) [35], Польше (7,3%) [37], Испании (0,01%) [29], Швейцарии (около 1%) [13], в США (1,5%) [24], Франции 2,3% [30].
Мутация c.101T>C выявлена в трех семьях с НСНТ из РБ. У одного индивида из выборки больных, башкира по этнической принадлежности, идентифицирована замена c.101T>C в гомозиготном состоянии (рис. 3Б), у второго пациента, метиса (русский/татарин), в компаунд гетерозиготном состоянии с полиморфным вариантом с.79A>G (рис. 3А) в гене GJB2 и у третьего пациента русской этнической принадлежности в сочетании с мутацией q.-53-2A>G в гене SLC26A5 . У всех трех пациентов наблюдалась III степень тугоухости. Это не противоречит литературными данными, что мутация q.-53-2A>G идентифицирована у пациента, русского по этнической принадлежности, т.к. трансверсия q.-53-2A>G в гене SLC26A5 встречается, в основном, в популяциях Европы и частота гетерозиготного носительства данной мутации у европейцев около 4%. В Эстонии мутация q.-53-2A>G выявлена с частотой 2,1% среди пациентов с НСНТ [34]. Частота мутации c.101T>C составила 1% у пациентов из РБ.
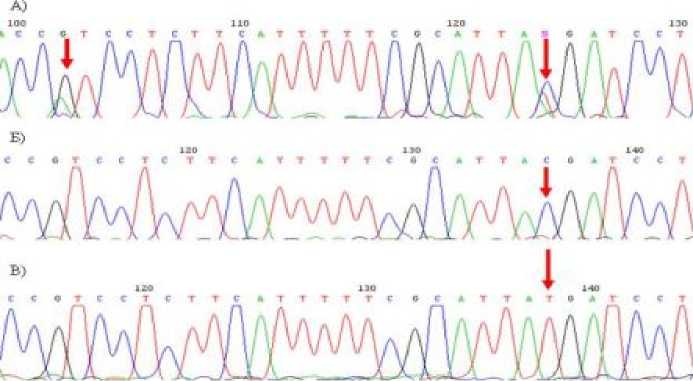
Рис. 3. Последовательность участка 2 экзона гена GJB2, содержащего мутацию c.101T>C в компаунд гетерозиготном состоянии с полиморфным вариантом с.79A>G (А); мутация c.101T>C в гомозиготном состоянии (Б) и нормальная последовательность гена GJB2 (В).
В заключение можно отметить, что хотя в этиологии и патогенезе заболевания остается много неясных аспектов, на сегодня известно, что примерно половина всех случаев врожденной глухоты имеет наследственное происхождение. Большинство случаев генетически детерминированной потери слуха наследуется по аутосомно-рецессивному типу.
Однако выраженная клиническая и генетическая гетерогенность заболевания, а также большая ассортативность браков между глухими и слабослышащими зачастую не позволяет точно выяснить механизм наследования дефекта звуковосприя-тия, а также приводит к повышению случаев болезни среди потомков пробанда.
Большинство детей с врожденными дефектами слуха имеют слышащих родителей, между тем отсутствие сведений о родственниках, имеющих нарушения звуковосприятия, не исключает возможность генетической природы заболевания. Как следствие, правильное консультирование семей таких пациентов практически невозможно без применения ДНК-диагностики.
Список литературы Молекулярно-генетический анализ генов GJB2, SLC26A4 и SLC26A5 у больных с наследственными нарушениями слуха из Республики Башкортостан
- Albert S., Blons H., Jonard L. et. al. SLC26A4 gene is frequently involved in nonsyndromic hearing impairment with enlarged vestibular aqueduct in Caucasian populations//Eur. J. Hum. Genet. 2006. № 14. P. 773-779.
- Bicego M., Beltramello M., Melchionda S. et. al. Pathogenetic role of the deafness-related M34T mutation of Cx26//Hum. Mol. Genet. 2006. V. 15. № 17. P. 2569-2587.
- Bors A., Andrikovics H., Kalmar L. et al. Frequencies of two common mutations (c.35delG and c.167delT) of the connexin 26 gene in different populations of Hungary//Intern. J. Mol. Med. 2004. № 14. P. 1105-1108.
- Conne B.W. Sensory Transduction//Medical Physiology: A Cellular and Molecular Approach. Philadelphia, 2002. P. 325-358.
- Connexin-Deafness Homepage, http://davinci.crg.es/deafness/
- Dallos P. The active cochlea//J. Neurosci. 1992. V. 12. P. 4575-4585.
- Dahl H.H., Tobin S.E., Poulakis Z. et al. The contribution of GJB2 mutations to slight or mild hearing loss in Australian elementary school children//J. Med. Genet. 2006. V. 43 (11). P. 850-855.
- D'Andrea P., Veronesi V., Bicego M. Hearing loss: frequency and functional studies of the most common connexin26 alleles//Biochem Biophys Res. Comm. 2002. V. 296 (3). P. 685-691.
- Eisen M.D., Ryugo D.K. Hearing molecules: contributions from genetic deafness//Cell. Mol. Life Sci. 2007. № 64. P. 566-580.
- Everett L.A., Glaser B. et al. Pendred syndrome is caused dy mutations in a putative sulphate transporter gene (PDS)//Nat. Genet. 1997. V. 17. P. 411-422.
- Fraser G.R. Association of congenital deafness with goitre (Pendred’s syndrome): a study of 207 families//Ann. Hum. Genet. 1965. V. 28. P. 201-249.
- Griffith A.J., Chowdhry A.A., Kurima K. et al. Autosomal recessive nonsyndromic neurosensory deafness at DFNB1 not associated with the compound-heterozygous GJB2 (connexin 26) genotype M34T/167delT//Am. J. Hum. Genet. 2000. V. 67. P. 745-749.
- Gürtler N., Kim Y., Mhatre A. et al. GJB2 mutations in the Swiss hearing impaired//Ear Hear. 2003. V. 24 (5). P. 440-447.
- Janecke A.R., Hirst-Stadlmann A., Günther B. et al. Progressive hearing loss, and recurrent sudden sensorineural hearing loss associated with GJB2 mutations-phenotypic spectrum and frequencies of GJB2 mutations in Austria//Hum. Genet. 2002. V. 111 (2). P. 145-53.
- Kelley P.M., Harris D.J., Comer B.C. et al. Novel mutations in the connexin 26 gene (GJB2) that cause autosomal recessive (DFNB1) hearing loss//Am. J. Hum. Genet. 1998. V. 62. P. 792-799.
- Kelsell D.P., Dunlop J., Stevens H.P. et al. Connexin 26 gene mutations in hereditary non-syndromic sensorineural deafness//Nature. 1997. V. 387. P. 80-83.
- Kelsell D.P., Wilgoss A.L., Richard G. et al. Connexin mutations associated with palmoplantar keratoderma and profound deafness in a single family//Eur. J. Hum.Genet. 2000. V. 8 (6). P. 469-72.
- Kenna M.A., Feldman H.A., Neault M.W. et al. Audiologic phenotype and progression in GJB2 (Connexin 26) hearing loss//Arch. Otolaryngol Head Neck Surg. 2010. V. 136 (1). P. 81-87.
- Löppönen T., Väisänen M.L., Luotonen M. et al. Connexin 26 mutations and nonsyndromic hearing impairment in northern Finland//Laryngoscope. 2003. V. 113 (10). P. 1758-1763.
- Mazzoli M., Van Camp G., Newton V. et al. Recommendations for the description of genetic and audiological data for families with nonsyndromic hereditary hearing impairment//Composed by the GENDEAF study group on genotype phenotype correlations. 2003. P. 1-3.
- Morell R.J., Kim H.J., Hood L.J. Mutations in the connexin 26 gene (GJB2) among Ashkenazi Jews with nonsyndromic recessive deafness//N. Engl. J. Med. 1998. V. 339 (21). P. 1500-1505.
- Morton N.E., Ann N.Y. Genetic epidemiology of hearing impairment//Acad. Sci. 1991. № 630. P. 16-31.
- Nance W.E. The genetic of deafness//MRDD Res. Rev. 2003. V. 9. P. 109-119.
- Pandya A., Arnos K.S., Xia X.J. et al. Frequency and distribution of GJB2 (connexin 26) and GJB6 (connexin 30) mutations in a large North American repository of deaf probands//Genet. Med. 2003. V. 5 (4). P. 295-303.
- Park H.J., Lee S.J., Jin H.S. et al. Genetic basis of hearing loss associated with enlarged vestibular aqueducts in Koreans//Clin. Genet. 2004. V. 67. P. 160-165.
- Park H.J., Shaukat S., Liu X.Z. et al. Origins and frequencies of SLC26A4 (PDS) mutations in east and south Asians: global implications for the epidemiology of deafness//J. Med. Genet. 2003. V. 40. P. 242-248.
- Petersen M.B., Willems P.J. Non-syndromic, autosomal-recessive deafness//Clin. Genet. 2006. № 69. P. 371-392.
- Primignani P., Trotta L., Castorina P. et al. Analysis of the GJB2 and GJB6 genes in Italian patients with nonsyndromic hearing loss: frequencies, novel mutations, genotypes, and degree of hearing loss//Genet. Test Mol. Biomarkers. 2009. V. 13 (2). P. 209-217.
- Rabionet R., Gasparini P., Estivill X. Molecular genetics of hearing impairment due to mutations in gap junction genes encoding beta connexins//Hum. Mutat. 2000. V. 16. P. 190-202.
- Roux A.F., Pallares-Ruiz N., Vielle A. et al. Molecular epidemiology of DFNB1 deafness in France//BMC Med. Genet. 2004. V. 5. P. 1-10.
- Smith S.D., W.J. Kimberling, Schaefer G.B. et al. Medical genetic evaluation for the etiology of hearing loss in children//J. Commun. Disord. 1998. V. 31. P. 371-388.
- Stephens D. Audiological terms//Definitions, protocols & guidelines in genetic hearing impairment. Whurr publishers, 2001.
- Tan, W., Zhang Y., Chang Q. et al. Connexin29 is highly expressed in cochlear Schwann cells, and it is required for the normal development and function of the auditory nerve of mice//J. Neurosci. 2006. V. 26. P. 1991-1999.
- Teek R., Oitmaa Eneli E., Kruustuk K. et al. Splice variant IVS2-2A>G in the SLC26A5 (Prestin) gene in five Estonian families with hearing loss//Intern. J. Pediatric Otorhinolaryngology. 2009. № 73. P. 103-107.
- Tóth T., Kupka S., Haack B. et al. GJB2 mutations in patients with non-syndromic hearing loss from Northeastern Hungary//Hum. Mutat. 2004. V. 23 (6). P. 631-632.
- White T.W., Deans M.R., Kelsell D.P. et al. Connexin mutations in deafness//Nature. 1998. V. 394. P. 630-631.
- Wiszniewski W., Sobieszczanska-Radoszewska L., Nowakowska-Szyrwinska E. et al. High frequency of GJB2 gene mutations in Polish patients with prelingual nonsyndromic deafness//Genet. Test. 2001. V. 5 (2). P. 147-148.
- Zheng J., Shen W., He D.Z.Z et al. Prestin is the motor protein of cochlear outer hair cells//Nature. 2000. V. 405. P. 149-155.
