Multiple Features Based Approach to Extract Bio-molecular Event Triggers Using Conditional Random Field

Author: Amit Majumder
Journal: International Journal of Intelligent Systems and Applications(IJISA) @ijisa
Article in issue: 12 vol.4, 2012.
Free access
The purpose of Biomedical Natural Language Processing (BioNLP) is to capture biomedical phenomena from textual data by extracting relevant entities, information and relations between biomedical entities (i.e. proteins and genes). In general, in most of the published papers, only binary relations were extracted. In a recent past, the focus is shifted towards extracting more complex relations in the form of bio-molecular events that may include several entities or other relations. In this paper we propose an approach that enables event trigger extraction of relatively complex bio-molecular events. We approach this problem as a detection of bio-molecular event trigger using the well-known algorithm, namely Conditional Random Field (CRF). We apply our experiments on development set. It shows the overall average recall, precision and F-measure values of 64.27504%, 69.97559% and 67.00429%, respectively for the event detection.
BioNLP, Conditional Random Field (CRF), Event, Event Trigger, Template
Short address: https://sciup.org/15010344
IDR: 15010344
Text of the scientific article Multiple Features Based Approach to Extract Bio-molecular Event Triggers Using Conditional Random Field
Published Online November 2012 in MECS
-
I. Introduction
The past history of text mining (TM) shows the great success of different evaluation challenges based on carefully curated resources, such as those organized in the MUC (Chinchor, 1998), TREC (Voorhees, 2007) and ACE (Strassel et al., 2008) events. All these shared tasks have significantly contributed to the progress of their respective fields. This has also been similar for bio-text mining (bio-TM). Some of the bio-text mining evaluation challenges include the TREC Genomics track (Hersh et al., 2007), JNLPBA (Kim et al., 2004), LLL (N´edellec, 2005), and BioCreative (Hirschman et al., 2007). The first two shared tasks addressed the issues of bio-information retrieval (bio-IR) and bioNamed Entity Recognition (bio-NER), respectively. The last two evaluation campaigns were associated with the bio-information extraction (bio-IE). These two addressed the issues of seeking relations between biomolecules. With the emergence of NER systems with performance capable of supporting practical
applications, the recent interest of the bio-TM community is shifting toward IE.
Relations among biomedical entities (i.e. proteins and genes) are important in understanding biomedical phenomena and must be extracted automatically from a large number of published papers. Most researchers in the field of Biomedical Natural Language Processing (BioNLP) have focused on extracting binary relations, including protein–protein interactions (PPIs) such as LLL and BioCreative challenges. Binary relations are not sufficient for capturing biomedical phenomena in detail, and there is a growing need for capturing more detailed and complex relations. For this purpose, two large corpora, BioInfer and GENIA, have been proposed.
Similarly to previous bio-text mining challenges (e.g., LLL and BioCreative), the BioNLP’09 Shared Task (also addressed bio-IE, but it tried to look one step further toward finer-grained IE. The difference in focus is motivated in part by different applications envisioned as being supported by the IE methods. For example, BioCreative aims to support curation of PPI databases such as MINT (Chatr-aryamontri et al., 2007), for a long time one of the primary tasks of bioinformatics. The BioNLP’09 shared task contains simple events and complex events. Whereas the simple events consist of binary relations between proteins and their textual triggers, the complex events consist of multiple relations among proteins, events, and their textual triggers. Bindings can represent events among multiple proteins, and regulations can represent causality relations between proteins and events. These complex events are more informative than simple events, and this information is important in modelling biological systems, such as pathways. The primary goal of BioNLP-09 shared task was aimed to support the development of more detailed and structured databases, e.g. pathway (Bader et al., 2006) or Gene Ontology Annotation (GOA) (Camon et al., 2004) databases, which are gaining increasing interest in bioinformatics research in response to recent advances in molecular biology.
In the present paper, we propose a system that enables the extraction of bio-molecular events from the medical literature. The main goal of event extraction is to detect the bio-molecular events from the texts and to classify them into nine predefined classes, namely gene expression, transcription, protein catabolism, phosphorylation, localization, binding, regulation, positive regulation and negative regulation. We approach the problem from a supervised machine learning perspective based on Conditional Random Field (CRF) that makes use of statistical and linguistic features that represent various morphological, syntactic and contextual information of the candidate bio-molecular trigger words.
In this paper, I describe the proposed approach for event word extraction, task definition, datasets description and experimental results followed by conclusion and future scope.
-
II. Proposed Approach for Event Word Extraction
-
2.1 Conditional Random Field
Conditional Random Field (CRF) (Lafferty et al., 2001) is an undirected graphical model, which is a special case of which corresponds to conditionally trained probabilistic finite state automata. The main advantage of CRF comes from that it can relax the assumption of conditional independence of the observed data often used in generative approaches, an assumption that might be too restrictive for a considerable number of object classes. Additionally, CRF avoids the label bias problem.
In this section we describe our proposed approach for event extraction that involves identification of bio-molecular events from the texts and classification of them into some predefined categories of interest. We approach this problem from the supervised machine learning perspective and use Conditional Random Field (CRF). We use a diverse set of features varying from morphological, syntactic and local as well as global context information.
In our approach we have converted the multi-word events into single-word event. To do this, first, we have made a list of event words consisting of single word event and multi-word event from the training set. Based on this list, all multi-word events are converted to single word event using “_”. For example, if a multi-word event is “Gene expression”, then this is converted to Gene_expression. In development set, the multi-word events which are not in this list can not be converted to single-word event. In this case, the first word is considered as event word for getting evaluation result.
CRF is used to calculate the conditional probability of values on designated output nodes given values on other designated input nodes. The conditional probability of a state sequence
S =< s 1, s 2,
S T >
, „ ■■ O =< O1,o2,.....,OT) ■ given an observation sequenceis calculated as:
TK
Pл(s | o) = — exp(^ ^ Xkfk(st —1, st, o, t)) Zo 1=11=1
where, fk(st 1, st, o, t) is a feature function whose weight Xk is to be learned via training. The values of the feature functions may range between — ^.....+ but typically they are binary. To make all conditional probabilities sum up to 1, we must calculate the normalization factor,
TK
Z 0 = Z s exp( Z Z X k f k ( s t — 1 , s t , o , t )) t = 1 k = 1 , (2)
This, as in HMMs, can be obtained efficiently by dynamic programming.
Here, the CRF parameters are optimized using Limited-memory BFGS[16], a quasi-Newton method that is significantly more efficient, and results in only minor changes in accuracy due to changes in ^ . CRFs generally can use real-valued functions but it is often required to incorporate the binary valued features. A feature function fk ( s t 1 , s t , o , t ) has a value of 0 for most cases and is only set to 1, when t 1 , t are certain states and the observation has certain properties. We use the C++ based CRF++ package 1 , a simple, customizable, and open source implementation of CRF for segmenting /labeling sequential data.
-
2.2 Features for Event Extraction
In our work, we define and use the following set of features for event extraction. All these features are automatically extracted from the training datasets without using any additional domain dependent resources and/or tools. We use CRF++ to generate model by running the tool on training data set.
As CRF++ is designed as a general purpose tool, you have to specify the feature templates in advance. This file describes which features are used in training and testing.
Each line in the template file denotes one template. In each template, special macro %x[row,col] will be used to specify a token in the input data. row specfies the relative position from the current focusing token and col specifies the absolute position of the column. In the following, %x[0,n] represents a n-th feature of a feature vector of current token.
List of features:
%x[0,0]: word
%x[0,1]: root word
%x[0,2]: POS of the word
%x[0,3]: POS of the word in BOI format.
%x[0,4]: named entity tag
%x[0,5]: whether protein or others
%x[0,6]: POS of previous token
%x[0,7]: POS of previous to previous token
%x[0,8]: distance from nearest protein
%x[0,9]: name of nearest protein
%x[0,10]: root word of name of nearest protein %x[0,11] to %x[0,20]: ten Boolean features.
To find this feature, frequencies of event words in training set are found. On the basis of frequencies, these words are sorted in descending order and top ten words are taken into consideration to create ten Boolean features.
%x[0,21]: dependency path from nearest protein in that sentence
%x[0,22]: boolean feature ("true" if dependency label between token and its child is OBJ)
%x[0,23]: length of dependency path
%x[0,24] to %x[0,33]: frequency of named-entities in sliding window
%x[0,34] to %x[0,41]: Four prefix features and four suffix features
%x[0,42] to %x[0,51]: previous five and next five words
%x[0,52] to %x[0,55]: previous two (POS and NE pair)
%x[0,56] to %x[0,59]: next two (POS and NE pair)
%x[0,60]: boolean feature.
considering was centre we define a context window as: context-window-size=2*context-size+1. When the size exceeds the length of the sentence, we added some slots and fill it by the class labels “Other-than-NEs” (denoted by O). For word w, a feature vector of length 5 is defined. Depending upon the value of K, the corresponding feature fires. The value is set equal to the number of NEs within the contexts of “context-window-size”. For example, for K=1, the entire sentence is considered (i.e., context-size=L). For the first word of the sentence, the context window is equal to more than twice (i.e., 2*context-size+1) of the sentence length. For K=2, the context-size is half of the sentence length. Again, centring the word w we define a context of double length by filling the preceding empty slots with O. The feature value is equal to the number of NEs within this window.
Dependency path (Feature):
Dependency relations of the path from the nearest protein are used as the features. Previous approaches use both the words and the dependency relation types to represent the paths (Bunescu and Mooney, 2007; Erkan et al., 2007). Consider the dependency tree in Figure 1. The path from “phosphorylation” to “CD40” is “nsubj inhibits acomp binding prep to domain num”. Due to the large number of possible words, use of these words on the paths may lead to data sparsity problems, and in turn to poor generalization. Suppose we have a sentence with similar semantics, where the synonym word “prevents” is used instead of “inhibits”. If we use the words on the path to represent the path feature, we end up with two different paths for the two sentences that have similar semantics. Therefore, in this work we use only the dependency relation types among the words to represent the paths. For example, the path feature extracted for the (phosphorylation, CD40) negative trigger/participant pair is “nsubj acomp prep to num” and the path feature extracted for the (phosphorylation, TRAF2) positive trigger/participant pair is “prep of”.
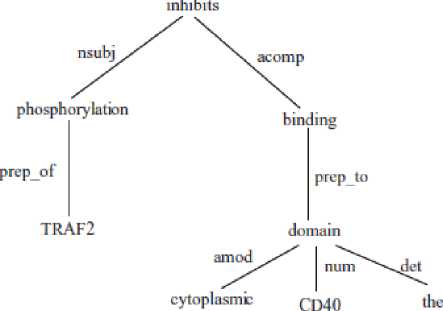
Figure 1: The dependency tree of the sentence "The phosphorylation ofTRAF2 inhibits binding to the CD40 cytoplasmic domain."
|
CRF++ takes training dataset and one template to generate model file. This model file is applied on test dataset (or on development dataset) to generate output. The following template has been used by CRF++ tool generate the model file. |
U33:%x[0,50] U34:%x[0,35]/%x[0,1] U35:%x[0,36]/%x[0,1] U36:%x[0,37]/%x[0,1] U37:%x[0,38]/%x[0,1] |
|
Template-t: |
U38:%x[0,39]/%x[0,1] |
|
# Unigram |
U39:%x[0,40]/%x[0,1] |
|
U0:%x[0,0] |
U40:%x[0,41]/%x[0,1] |
|
U1:%x[0,1] |
U41:%x[0,42]/%x[0,1] |
|
U2:%x[0,2] |
U42:%x[0,43]/%x[0,1] |
|
U3:%x[0,3] |
U43:%x[0,44]/%x[0,1] |
|
U4:%x[0,6] |
U44:%x[0,45]/%x[0,1] |
|
U5:%x[0,7] |
U45:%x[0,46]/%x[0,1] |
|
U6:%x[0,8] |
U46:%x[0,47]/%x[0,1] |
|
U7:%x[0,9] |
U47:%x[0,48]/%x[0,1] |
|
U8:%x[0,10] |
U48:%x[0,49]/%x[0,1] |
|
U9:%x[0,11] |
U49:%x[0,50]/%x[0,1] |
|
U10:%x[0,12] |
U50:%x[0,51] |
|
U11:%x[0,13] |
U51:%x[0,52] |
|
U12:%x[0,14] |
U52:%x[0,53] |
|
U13:%x[0,15] |
U53:%x[0,54] |
|
U14:%x[0,16] |
U54:%x[0,55] |
|
U15:%x[0,17] |
U55:%x[0,56] |
|
U16:%x[0,18] |
U56:%x[0,57] |
|
U17:%x[0,19] |
U57:%x[0,58] |
|
U18:%x[0,20] |
U58:%x[0,59] |
|
U19:%x[0,21] |
U59:%x[0,60] |
|
U20:%x[0,22] |
U60:%x[0,44]/%x[0,45]/%x[0,46]/%x[0,47]/%x[0,4 |
|
U21:%x[0,23] |
8] |
|
U22:%x[0,39] |
U61:%x[0,44]/%x[0,45]/%x[0,46]/%x[0,47] |
|
U23:%x[0,40] |
U62:%x[0,44]/%x[0,45]/%x[0,46] |
|
U24:%x[0,41] |
U63:%x[0,44]/%x[0,45] |
|
U25:%x[0,42] |
U64:%x[0,49]/%x[0,50]/%x[0,51]/%x[0,52]/%x[0,5 |
|
U26:%x[0,43] |
3] |
|
U27:%x[0,44] |
U65:%x[0,49]/%x[0,50]/%x[0,51]/%x[0,52] |
|
U28:%x[0,45] |
U66:%x[0,49]/%x[0,50]/%x[0,51] |
|
U29:%x[0,46] |
U67:%x[0,49]/%x[0,50] |
|
U30:%x[0,47] |
U68:%x[0,0]/%x[0,5] |
|
U31:%x[0,48] |
U69:%x[0,8]/%x[0,1] |
|
U32:%x[0,49] |
U70:%x[0,9]/%x[0,1] U71:%x[0,10]/%x[0,1] |
U72:%x[0,9]/%x[0,1]
# Bigram
B
-
III. Task Definition, Datasets and Experimental Results
-
3.1 Task definition:
-
The BioNLP Shared Task focuses on extraction of bio-events particularly on proteins or genes (Proteins and gene are not distinguished). To concentrate efforts on the novel aspects of the extraction task, it is assumed that the protein recognition has been already performed, and the shared task begins with a gold standard set of proteins annotations. The shared task is designed to address a semantically rich IE problem as a whole, but divided into three subtasks to allow separate evaluation of the performance for different aspects of the problem.
Task 1. Core event extraction
This task is to identify events concerning the given proteins. This task involves event trigger detection, event typing, and primary argument recognition.
e.g.) phosphorylation of TRAF2
-
— > (Type: Phosphorylation, Theme: TRAF2)
Task 2. Event enrichment (optional)
This task is to find secondary argumentsents of events that further specify the event extracted by Task 1. This task involves the recognition of entities (other than proteins) and the assignment of these entities as event arguments.
e.g.) localization of beta-catenin into nucleus
—> (Type: Localization, Theme: beta-catenin, ToLoc: nucleus)
Task 3. Negation and speculation recognition (optional)
This task is to find negations and speculations regarding events extracted by Task 1.
e.g.) TRADD did not interact with TES2
—> (Negation (Type: Binding, Theme: TRADD, Theme:TES2))
We are working on task-1. Event word detection is part of task-1.
Example:
Consider the following sample text from biological domain.
Text: TRADD was the only protein that interacted with wild-type TES2 and not with isoleucine-mutated TES2.
Protein annotation (filename.a1) of the above text:
T1 Protein 0 5 TRADD
T2 Protein 58 62 TES2
T3 Protein 95 99 TES2
From the above text and protein annotation files, we have to generate the following event annotation corresponding to task-1.
Event annotation corresponding to Task 1:
T4 Binding 32 42 interacted
E1 Binding:T4 Theme:T1 Theme2:T2
E2 Binding:T4 Theme:T1 Theme2:T3
-
3.2 Datasets:
-
3.3 Experimental Results
The BioNLP-09 shared task datasets were prepared based on the GENIA event corpus. Training and development datasets were derived from the publicly available event corpus (Kim et al., 2008). The test set was obtained from an unpublished portion of the corpus. We present some statistics of the datasets in Table 1. The shared task organizers made some changes to the original GENIA event corpus. Irrelevant annotations were removed, and some new types of annotation were added to make the event annotation more appropriate.
Table 1: Statistics of the datasets
|
Dataset |
#abstracts |
#sentences |
#words |
#events |
|
Training |
800 |
7,449 |
176,146 |
8,597 |
|
Development |
150 |
1,450 |
33,937 |
1,809 |
|
Test |
260 |
2,447 |
57,367 |
3,182 |
The named entity (NE) annotation of the GENIA corpus has been somewhat controversial due to differences in annotation principles compared to other biomedical NE corpora. There is no distinction between proteins and genes in the widely applied GENETAG corpus (Tanabe et al., 2005), but in GENIA there were differences between these two. Such differences have caused significant inconsistency in methods and resources following different annotation schemes. In the GENIA corpus, appropriate revision of the original annotation was made to remove and/or reduce the inconsistency. Details can be found in Ohta et al. (2009).
We use CRF for training and testing. The system is tuned on the development data, and the results are reported using 3-fold cross validation. It is to be noted that due to the unavailability of gold-standard annotations we were unable to evaluate the system with the test dataset. The system is evaluated with the standard recall, precision and F-measure metrics. We followed the strict matching criterion, i.e. credit is given if and only if (1) the event types are the same and (2) the event triggers are the same.
Table 2: Evaluation results of event detection on the development set (we report in percentages)
|
Feature template |
recall |
precision |
F-measure |
|
Template-t given above |
64.27504 |
69.97559 |
67.00429 |
-
IV. Conclusion and Future Work
In this paper we have proposed a multiple features based approach to extract bio-molecular event triggers using Conditional Random Field that involves identification of complex bio-molecular events. We have used CRF that exploits various statistical and linguistic features in the forms of morphological, syntactic and contextual information of the candidate bio-molecular trigger words.
Overall evaluation results suggest that there is still the room for further improvement. We would like to investigate distinct and more effective set of features for event identification and to classify them according to predefined nine classes. We would also like to find out the arguments of the identified events. We also would like to try do our experiment with other tools, especially support vector machine, which may improve the experimental result.
References Multiple Features Based Approach to Extract Bio-molecular Event Triggers Using Conditional Random Field
- Hyoung-Gyu Lee, Han-Cheol Cho, Min-Jeong Kim Joo-Young Lee, Gumwon Hong, Hae-Chang Rim. A Multi-Phase Approach to Biomedical Event Extraction. In BioNLP ’09: Proceedings of the Workshop on BioNLP, 107-110.
- Arzucan O¨ zgu¨ r, Dragomir R. Radev. Supervised Classification for Extracting Biomedical Events. BioNLP ’09: Proceedings of the Workshop on BioNLP, 111-114.
- Pyysalo S, Ginter F, Heimonen J, Bj¨orne J, Boberg J, J¨arvinen J, Salakoski T, BioInfer: A corpus for information extraction in the biomedical domain, BMC Bioinformatics 8:50, 2007.
- Kim J-D, Ohta T, Tsujii J, Corpus annotation for mining biomedical events from literature, BMC Bioinformatics 9:10, 2008.
- Kim J-D, Ohta T, Pyysalo S, Kano Y, Tsujii J, Overview of BioNLP’09 shared task on event extraction, in BioNLP ’09: Proceedings of the Workshop on BioNLP, pp. 1–9, 2009.
- Nancy Chinchor. 1998. Overview of MUC-7/MET-2. In Message Understanding Conference (MUC-7) Proceedings.
- Asif Ekbal, Amit Majumder, Mohammad Hasanuzzaman and Sriparna Saha. Supervised Machine Learning Approach for Bio-molecular Event Extraction in Swarm, Evolutionary, and Memetic Computing (SEMCO), 2011
- Ellen Voorhees. 2007. Overview of TREC 2007. In the Sixteenth Text REtrieval Conference (TREC 2007) Proceedings.
- Sriparna Saha, M. Hasanuzzaman, Amit Majumder, Asif Ekbal. Bio-molecular event extraction using Support Vector Machine in Third International Conference on Advanced Computing (ICoAC), 2011
- Stephanie Strassel, Mark Przybocki, Kay Peterson, Zhiyi Song, and Kazuaki Maeda. 2008. Linguistic Resources and Evaluation Techniques for Evaluation of Cross-Document Automatic Content Extraction. In Proceedings of the 6th International Conference on Language Resources and Evaluation (LREC 2008).
- Lynette Hirschman, Martin Krallinger, and Alfonso Valencia, editors. 2007. Proceedings of the Second BioCreative Challenge Evaluation Workshop. CNIO Centro Nacional de Investigaciones Oncol´ogicas.
- Jin-Dong Kim, Tomoko Ohta, Yoshimasa Tsuruoka, Yuka Tateisi, and Nigel Collier. 2004. Introduction to the bio-entity recognition task at JNLPBA. In Proceedings of the International Joint Workshop on Natural Language Processing in Biomedicine and its Applications (JNLPBA), pages 70–75.
- Claire N´edellec. 2005. Learning Language in Logic -Genic Interaction Extraction Challenge. In J. Cussens and C. N´edellec, editors, Proceedings of the 4th Learning Language in Logic Workshop (LLL05), pages 31–37.
- Andrew Chatr-aryamontri, Arnaud Ceol, Luisa Montecchi Palazzi, Giuliano Nardelli, Maria Victoria Schneider, Luisa Castagnoli, and Gianni Cesareni. 2007. MINT: the Molecular INTeraction database. Nucleic Acids Research, 35(suppl 1):D572–574.
- Gary D. Bader, Michael P. Cary, and Chris Sander. 2006. Pathguide: a Pathway Resource List. Nucleic Acids Research., 34(suppl 1):D504–506.
- Evelyn Camon, Michele Magrane, Daniel Barrell, Vivian Lee, Emily Dimmer, John Maslen, David Binns, Nicola Harte, Rodrigo Lopez, and Rolf Apweiler. 2004. The Gene Ontology Annotation (GOA) Database: sharing knowledge in Uniprot with Gene Ontology. Nucl. Acids Res., 32(suppl 1):D262–266.
- Jin-Dong Kim, Tomoko Ohta, and Jun’ichi Tsujii. 2008. Corpus annotation for mining biomedical events from lterature. BMC Bioinformatics, 9(1):10.
- Marie-Catherine de Marneffe, Bill MacCartney, and Christopher D. Manning. 2006. Generating Typed Dependency Parses from Phrase Structure Parses. In Proceedings of the Fifth International Conference on Language Resources and Evaluation (LREC’06), pages 449–454.
- Lorraine Tanabe, Natalie Xie, Lynne Thom, Wayne Matten, and John Wilbur. 2005. Genetag: a tagged corpus for gene/protein named entity recognition. BMC Bioinformatics, 6(Suppl 1):S3.
- Sampo Pyysalo, Tomoko Ohta, Jin-Dong Kim, and Jun’ichi Tsujii. 2009. Static Relations: a Piece in the Biomedical Information Extraction Puzzle. In Proceedings of Natural Language Processing in Biomedicine (BioNLP) NAACL 2009 Workshop, 1-9.
- Tomoko Ohta, Jin-Dong Kim, Sampo Pyysalo, and Jun’ichi Tsujii. 2009. Incorporating GENETAG-style annotation to GENIA corpus. In Proceedings of Natural Language Processing in Biomedicine (BioNLP) NAACL 2009 Workshop, 1-9.
- Gu¨nes¸ Erkan, Arzucan O¨ zgu¨r, and Dragomir R. Radev. 2007. Semi-supervised classification for extracting protein interaction sentences using dependency parsing. In Proceedings of EMNLP, pages 228–237.
- R. C. Bunescu and R. J. Mooney, 2007. Text Mining and Natural Language Processing, Chapter Extracting Relations from Text: From Word Sequences to Dependency Paths, pages 29–44, Springer.
- Lafferty, J., McCallum, A., Pereira, F. 2001 Conditional Random Fields: Probabilistic Models for Segmenting and Labeling Sequence Data. Proceedings of 18th International Conference on Machine Learning, pp.282-289.
- Sha, F., Pereira, F. 2003. Shallow Parsing with Conditional Random Fields. Proceedings of HLT-NAACL.
