Мутация гена UGT1A1 как маркер высокого риска возникновения синдрома Жильбера: научно-прикладные аспекты

Автор: Волков А.Н., Цуркан Е.В.
Журнал: Анализ риска здоровью @journal-fcrisk
Рубрика: Медико-биологические аспекты оценки воздействия факторов риска
Статья в выпуске: 2 (26), 2019 года.
Бесплатный доступ
Синдром Жильбера является распространенной мультифакториальной патологией с высокой степенью генетической детерминированности. Основной этиологический фактор заболевания - снижение активности печеночного фермента УДФ-глюкуронилтрансферазы А1 вследствие мутаций в гене UGT1A1. Нарушение функции печени становится причиной диспептических явлений и сопутствующих острых и хронических заболеваний пищеварительной системы. Целью работы явилось обоснование необходимости и возможности осуществления массового обследования населения на синдром Жильбера с использованием молекулярно-генетического анализа гена UGT1A1. Проведено молекулярно-генетическое исследование маркера rs8175347 гена UGT1A1 среди 132 жителей Кемеровской области (популяционная выборка), а также среди 71 пациента с подозрением на наличие синдрома Жильбера (клиническая выборка). В популяционной выборке частота мутантного генотипа *28/*28 гена UGT1A1, ассоциированного с синдромом Жильбера, составила 13,6 %, что сопоставимо с ранее опубликованными данными...
Синдром жильбера, удф-глюкуронилтрансфераза а1, мутации в гене, генотип, молекулярно-генетическое исследование, генодиагностика, udp-glucuronosyltransferase а1
Короткий адрес: https://sciup.org/142220674
IDR: 142220674 | УДК: 616-079.3: | DOI: 10.21668/health.risk/2019.2.14
UGT1A1 gene mutation as a marker indicating there is a high risk of Gilbert's syndrome: theoretical and applied aspects
Gilbert's syndrome is a widely spread multi-factor pathology which is to a great extent genetically determined. Its basic etiological factor is lower activity of a liver enzyme, UDP-glucuronosyltransferase A1, caused by mutations in UGT1A1 gene. Functional disorders in the liver cause dyspepsia and concurrent acute and chronic diseases in the digestive system. The research goal was to substantiate the necessity and possibility to conduct mass examinations of population with molecular and genetic analysis of UGT1A1 gene in order to reveal Gilbert's syndrome. The authors performed molecular and genetic examination of UGT1A1 gene rs8175347 marker in 132 people living in Kemerovo region (population sampling) as well as in 71 patients who were supposed to have Gilbert's syndrome (clinical sampling). Frequency of *28/*28 mutant genotype of UGT1A1 gene associated with Gilbert's syndrome amounted to 13.6 % in the population sampling and it is quite consistent with previously published data...
Текст научной статьи Мутация гена UGT1A1 как маркер высокого риска возникновения синдрома Жильбера: научно-прикладные аспекты
Волков Алексей Николаевич – кандидат биологических наук, доцент (e-mail: ; тел. 8 (3842) 734-856; ORCID: .
Цуркан Елена Владимировна – биолог (тел. 8 (3842) 396-023; ORCID: .
денных с гемолитическими состояниями синдром Жильбера существенно повышает риск осложнений и неонатальной гибели [2].
Биохимическая и генетическая основа СЖ в настоящее время однозначно установлена. Снижение активности УДФ-глюкуронилтрансферазы А1 при этом заболевании определяется либо изменением уровня экспрессии соответствующего гена UGT1A1 , либо структурными модификациями самого фермента. В первом случае обычно обнаруживается изменение числа динуклеотидных повторов TA в промоторной области гена (полиморфный маркер rs8175347 ). Так, если аллель «дикого типа» *1 характеризуется шестью тандемными повторами TА , то при мутациях, ассоциированных с СЖ, их число увеличивается до 7 (аллель *28 ) или 8 (аллель *37 ). Еще одна вариация промоторной области характеризуется уменьшением числа повторов TA до 5 (аллель *36 ) и приводит к повышению активности UGT1A1 без патологических проявлений [9, 10]. Установлено, что носители генотипа *1/*28 и, особенно, *28/*28 имеют в среднем более высокий уровень билирубина в сыворотке крови, чем гомозиготы * 1/*1 [11–13].
Данных о генетическом полиморфизме UGT1A1 , а также о его физиолого-биохимических проявлениях у населения России крайне мало [14, 15]. Кроме того, неизвестна частота синдрома Жильбера и объем группы риска по данной патологии у населения нашей страны. Это делает дополнительные популяционно-генетические исследования UGT1A1 необходимыми.
Актуальность и практическая значимость данной работы связаны с необходимостью разработки эффективных медико-диагностических алгоритмов выявления мультифакториальных патологий, таких как синдром Жильбера, на основе современных достижений генетики и смежных наук.
Цель исследования – обоснование необходимости и возможности осуществления массового обследования населения на синдром Жильбера с использованием молекулярно-генетического анализа гена UGT1A1 .
Материалы и методы. В ходе работы были сформированы две группы обследованных среди жителей Кемеровской области, принадлежащих к европеоидному типу (в основном русские). Популяционная группа состояла из сотрудников и пациентов Кемеровской областной клинической больницы обоих полов (68 женщин и 64 мужчины), в разное время проходивших диспансерное обследование в данном учреждении (табл. 1). Состояние здоровья обследованных не учитывалось. Данная выборка была использована для популяционно-генетического анализа полиморфного маркера rs8175347 гена UGT1A1 .
Клиническая выборка состояла из пациентов Кемеровской областной клинической больницы (38 женщин и 33 мужчины), ранее направленных в медико-генетическую консультацию учреждения
Таблица 1
Характеристика групп обследованных
|
Группа |
n |
Возрастные показатели, лет |
|||
|
М ± S.E. |
Me |
Mo |
min–max |
||
|
Популяционная |
132 |
36,4 ± 0,61 |
36 |
38 |
23–51 |
|
Клиническая |
71 |
34,3 ± 2,30 |
30 |
15 |
4–71 |
П р и м е ч а н и е : n – объем группы; M – среднее арифметическое значение; S.E . – стандартная ошибка среднего значения; Ме – медиана; Мо – мода; min–max – пределы варьирования.
на молекулярно-генетическое исследование в связи с предполагаемым синдромом Жильбера. Всем пациентам проведен анализ полиморфного участка rs8175347 гена UGT1A1 и получено заключение о гомозиготности по мутантному аллелю *28 , что является подтверждением предварительного диагноза. Данная группа позволила изучить некоторые возрастные аспекты манифестации и выявления СЖ.
У всех обследованных в стационарных условиях забирались образцы венозной крови с ЭДТА в качестве антикоагулянта. Выделение ДНК из цельной крови осуществляли на колонках «К-Сорб» производства НПК «Синтол» по инструкции производителя. ПЦР-амплификация полиморфного участка rs8175347 гена UGT1A1 проводилась с помощью коммерческого набора реагентов, разработанного ООО НПФ «Литех». Выявлялись аллель *1 (дикий тип, 6 ТА -повторов в промоторной области гена UGT1A ) и *28 (мутация, 7 ТА -повторов). Для детекции результата ПЦР использовался метод горизонтального электрофореза продуктов амплификации в 3%-ном агарозном геле с бромидом этидия в качестве красителя.
В ходе статистического анализа первичных данных для количественных переменных были рассчитаны основные выборочные показатели. Частоты аллелей и генотипов маркера rs8175347 гена UGT1A1 вычислялись как доли от их общего количества в выборке. Сравнение распределений качественных переменных осуществлялось с помощью критерия χ² в программе Statistica 6.0. Отличие считали достоверным при p <0,05.
Результаты и их обсуждение. Обсуждая генетические факторы риска СЖ, прежде всего следует оценить распространенность мутаций в гене UGT1A1 , ассоциированных с данной патологией. В исследованной нами популяционной выборке европеоидов Кемеровской области преобладающим генотипическим вариантом был *1/*1 (47,0 %) (табл. 2). Частота минорного аллеля *28 , связанного с патологическим состоянием, составила 33,3 %. При этом доля наиболее редкого генотипа *28/*28 , ассоциированного с синдромом Жильбера, составила 13,6 %. Установленное соотношение генотипов достоверно не отличалось от ожидаемого в соответствии с равновесием Харди – Вайнберга (χ² = 0,674; p = 0,714).
Таблица 2
Популяционно-генетические особенности маркера rs8175347 гена UGT1A1 в различных европеоидных этносах
|
Этническая принадлежность |
Генотипы UGT1A1 ( rs8175347 ), % |
Частота аллеля *28 , % |
Источник |
||
|
*1/*1 |
*1/*28 |
*28/*28 |
|||
|
Русские |
47,0 |
39,4 |
13,6 |
33,3 |
Собственные данные |
|
Русские |
40,4 |
50,0 |
9,6 |
34,6 |
[14] |
|
Русские |
42,4 |
51,7 |
5,9 |
31,8 |
[15] |
|
Хорваты |
39,9 |
49,8 |
10,2 |
35,1 |
[16] |
|
Итальянцы |
43,9 |
39,8 |
16,3 |
36,2 |
[17] |
|
Голландцы |
44,2 |
43,7 |
11,9 |
33,7 |
[18] |
|
Испанцы |
40,0 |
51,0 |
9,0 |
34,5 |
[19] |
|
Европеоиды США |
46,6 |
43,1 |
10,0 |
31,6 |
[20] |
Это свидетельствует об отсутствии у какого-либо из генотипов выраженного дезадаптивного значения и отбора против данного генотипа в рассматриваемой популяции.
Выявленная нами частота патологического генотипа довольно высока, что требует подтверждения на основе других независимых исследований. Данных о популяционно-генетических особенностях UGT1A1 у жителей России крайне мало. Недавно были опубликованы результаты исследования полиморфизма UGT1A1 в выборке жителей Юга России (г. Ростов-на-Дону) [14]. Согласно полученным результатам, доля генотипа *28/*28 составила 9,6 %, а частота аллеля *28 даже превышала установленное нами значение. При этом следует отметить, что изученная выборка, строго говоря, не может считаться популяционной, так как была сформирована из онкобольных, страдающих колоректальным раком. В другом исследовании E.G. Shatalova et al. [15] установили частоту аллелей и генотипов rs8175347 у здоровых русских женщин. Частота гомозигот *28/*28 составила лишь 5,9 %, а доля аллеля *28 – 31,8 %.
Как показывает анализ других работ по изучению полиморфизма UGT1A1 , доля клинически значимого аллеля *28 среди европеоидов обычно превышает 30 %, но не достигает 37 % [16–20]. Это соответствует частоте гомозиготного генотипа *28/*28 , как правило, около 10 % или несколько больше. Такая частота минорного аллеля вплотную приближается к критическому значению, при котором доли гомозигот *1/*1 и гетерозигот практически совпадают. Поэтому в различных исследованных группах отмечалась общая закономерность – сопоставимые частоты двух генотипов, *1/*1 и *1/*28 , обычно с небольшим преобладанием варианта *1/*1 .
Можно заключить, что полученные нами данные в целом соответствуют результатам ранее проведенных исследований. В европеоидных этносах доля потенциальных или уже выявленных больных синдромом Жильбера, которые являются носителями генотипа *28/*28 гена UGT1A1 , обычно превышает 10 %.
Установленная в различных исследованиях высокая частота гомозиготных носителей мутации гена UGT1A1 требует разработки и внедрения в медицинскую практику специальных алгоритмов для массового обследования населения с целью раннего выявления групп повышенного риска СЖ на основе высокоточных маркеров патологии. Как было сказано ранее, симптомы синдрома Жильбера проявляются обычно с началом пубертатного возраста и существенно варьируются у разных пациентов в зависимости от специфического сочетания внешних воздействий. В такой ситуации традиционные диагностические методы оказываются недостаточно точными и способствуют увеличению сроков постановки диагноза.
Вместе с тем современные знания об этиологии синдрома Жильбера и роли мутаций в гене UGT1A1 позволяют предложить надежный алгоритм диагностики СЖ на основе молекулярно-генетических подходов. Своевременное выявление носителей мутантного генотипа *28/*28 , в идеале – еще на донозологи-ческом этапе, позволит скорректировать дальнейший образ жизни потенциального больного таким образом, чтобы патология не проявилась или нанесла минимальный ущерб его здоровью.
Для определения оптимального возраста проведения диагностического обследования нами была изучена выборка пациентов с диагнозом СЖ, установленном на основании клинических проявлений и выявления генотипа *28/*28 UGT1A1 (рисунок).
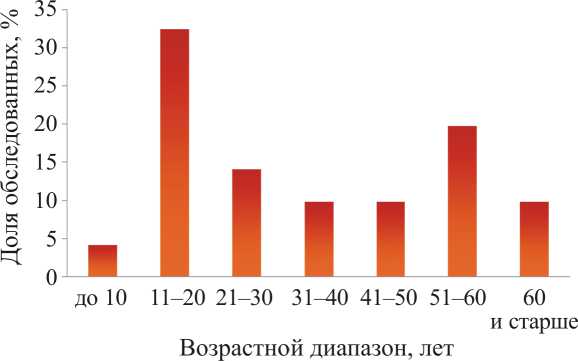
Рис. Возрастное распределение обследованных пациентов с синдромом Жильбера
Возраст первичной постановки диагноза СЖ может существенно варьироваться – от 4 лет до 71 года. В то же время более 30 % обследованных являются молодыми людьми (в возрасте до 20 лет), а модальное значение возраста в клинической выборке составило 15 лет. Это совпадает с ранее опубликованными наблюдениями, согласно которым основные проявления СЖ становятся заметными с началом полового созревания [3, 21]. Доля пациентов в старших возрастных группах несколько ниже. Второй заметный пик выявления СЖ отмечается среди лиц старше 50 лет, что может быть сопряжено со снижением функционального резерва организма в зрелом возрасте и суммарным эффектом патогенных факторов, действовавших в предыдущий период.
Как видно, среди обследованных всех возрастов сохраняются лица с впервые установленным диагнозом СЖ. Известно, что манифестация заболевания связана с индивидуальным сочетанием патогенных факторов, особенностями организма, образом жизни, диетой и пр. и оказывается непредсказуемой в онтогенетическом аспекте [1–3]. Тем не менее можно предположить, что наследственное снижение функции UGT1A1 в большинстве случаев рано или поздно проявится под влиянием эндогенных или экзогенных факторов. Кроме того, можно допустить наличие в популяции индивидуумов, имеющих те или иные проявления патологии на фоне носительства генотипа *28/*28 UGT1A1 и не обращавшихся в медицинские учреждения.
Как было отмечено ранее, поздняя постановка диагноза СЖ и отсутствие адекватной терапии могут сопровождаться высоким риском для здоровья пациента [5–8, 21]. На наш взгляд, оптимальными сроками для выявления СЖ является возраст до наступления пубертатного периода, когда симптомы патологии обычно еще отсутствуют. Такое обследование может быть реализовано в ходе диспансеризации школьников.
Согласно приказу Минздрава РФ «О порядке прохождения несовершеннолетними медицинских осмотров, в том числе при поступлении в образовательные учреждения и в период обучения в них»1, несовершеннолетние должны регулярно обследоваться в определенные возрастные периоды. Развернутое медицинское обследование школьники проходят в 7, 10, а также 14, 15, 16 и 17 лет. Примечательно, что действующая система обследования несовершеннолетних не предусматривает осмотр ребенка врачом-гастроэнтерологом, вследствие чего СЖ можно выявить только при появлении первых симптомов и жалоб со стороны больного.
Семилетний возраст обычно совпадает с поступлением ребенка в школу, что сопровождается появлением дополнительных факторов риска СЖ. К ним относятся физическое и психоэмоциональное напряжение, централизованное питание. Если до этого момента родители могут контролировать расписание дня, диетический режим и регулярность питания ребенка, то теперь это становится затруднительным. В ходе профилактического медицинского осмотра в возрасте 7 и 10 лет дети посещают ряд врачей. Кроме того, как и в прочие периоды, сдается кровь для общего анализа. Это могло бы стать оптимальным моментом для проведения диагностики СЖ на основе исследования мутаций гена UGT1A1 . При этом источником ДНК могли бы служить небольшие аликвоты цельной крови.
Методическая часть молекулярно-генетического исследования может быть реализована на осно- ве ранее описанных нами подходов с использованием отечественной инструментальной базы и тест-систем, что отвечает требованию импортозамеще-ния в области медицинских материалов и оборудования. При этом диагностическая процедура будет обладать низкой себестоимостью и простотой исполнения, что сделает ее доступной для массового применения [22].
На основании результатов, которые могут быть получены в ходе массового генетического обследования на синдром Жильбера, всех индивидуумов предлагается разделить на две группы. Носители генотипов *1/*1 и *1/*28 попадают в категорию «Условная норма», генотипа *28/*28 – «Группа риска по синдрому Жильбера». Дополнение «условная» необходимо, так как невозможно исключить наличие у этих обследованных иных, более редких мутаций в не изученных нами регионах гена UGT1A1 , которые также могут вызывать признаки СЖ. Вместе с тем доля таких лиц крайне мала, что было установлено в ходе ранее проведенных исследований в различных этнических группах [11, 20].
Категория «Группа риска по синдрому Жильбера» должна оказаться под пристальным вниманием врачей с обязательной постановкой на учет у врача-гастроэнтеролога и внесением генетической информации в амбулаторную карту ребенка. Поскольку предполагается генодиагностика среди детей, большинство из них могут не иметь признаков СЖ. Дальнейшая задача здравоохранения – информирование потенциальных пациентов и проведение эффективных профилактических мероприятий по недопущению манифестации СЖ. Это, прежде всего, относится к формированию у ребенка ответственного отношения к своему образу жизни и диете. Необходимо исключение факторов, провоцирующих развитие патологии. К ним относятся нервное и физическое напряжение, острые простудные заболевания, изобилие в диете низкокалорийной, острой, жареной пищи, а также голодание или нерегулярное питание и др.
Конкретные медицинские мероприятия в отношении пациента будут зависеть от особенностей проявления патологии. Помимо предусмотренных терапевтических мероприятий должен проводиться тщательный анализ причин, вызвавших манифестацию заболевания, с целью их дальнейшего исключения. Очевидно, знание о наличии генетической предрасположенности к синдрому Жильбера, соблюдение профилактических мер и своевременная адекватная терапия могут существенно улучшить качество жизни большинства лиц, оказавшихся в группе риска.
Выводы. Популяционно-генетическое исследование маркера rs8175347 гена UGT1A1 среди жителей Кемеровской области показало высокую час- тоту встречаемости мутантного генотипа *28/*28, ассоциированного с синдромом Жильбера (13,6 %). Очевидно, значительная часть населения включает потенциальных или уже выявленных больных СЖ. Это требует принятия мер по раннему выявлению лиц из группы генетического риска и применению в отношении их профилактических и терапевтических мероприятий.
Предложен алгоритм массового обследования на СЖ на донозологической стадии, в основу которого могут быть положены молекулярно-генетические технологии. В рамках развернутого медицинского обследования детей в возрасте 7 или 10 лет, предусмотренного действующими нормативными актами, предлагается проводить дополнительную генодиагностику мутаций UGT1A1 . Для ее осуществления можно использовать лабораторное оборудование и тест-системы отечественного производства, которые позволяют в кратчайшие сроки провести массовое обследование детей.
Полученные результаты могут быть учтены врачами соответствующих медицинских направлений для определения дальнейших мероприятий по профилактике и терапии синдрома Жильбера. Правильное определение или корректировка образа жизни, поведения, диеты позволит существенно улучшить качество жизни лиц с наследственной предрасположенностью к СЖ.
Предложенный алгоритм служит моделью трансляционной медицины в действии. Аналогичная схема может быть применена и в отношении других мультифакториальных заболеваний с изученной генетической компонентой и высокой наследственной детерминированностью. На основе подобных мероприятий, в идеале, должна быть сформирована система персонифицированной медицины, в фокусе которой находятся индивидуальные генетические и физиологические особенности пациента.
Финансирование . Исследование не имело спонсорской поддержки.
Список литературы Мутация гена UGT1A1 как маркер высокого риска возникновения синдрома Жильбера: научно-прикладные аспекты
- Strassburg C.P. Hyperbilirubinemia syndromes (Gilbert-Meulengracht, Crigler-Najjar, Dubin-Johnson, and Rotor syndrome)//Best Practice & Research Clinical Gastroenterology. -2010. -Vol. 24. -P. 555-571.
- Memon N. Inherited disorders of bilirubin clearance//Pediatr Res. -2016. -Vol. 79, № 3. -P. 378-386.
- Апостериорная ценность клинических и лабораторных проявлений синдрома Жильбера у детей/И.Н. Захарова, М.И. Пыков, З.В. Калоева, Л.А. Катаева, С.В. Шишкина, И.В. Бережная //Педиатрическая фармакология. -2011. -Т. 8, № 4. -С. 101-104.
- Дубровина Г.М., Ботвиньев О.К., Колотилина А.И. Сочетание синдрома Жильбера с заболеваниями желудочно-кишечного тракта//Российский журнал гастроэнтерологии, гепатологии, колопроктологии. -2014. -№ 3. -С. 13-21.
- Loci from a genome-wide analyses of bilirubin levels are associated with gallstone risk and composition/S. Buch, C. Schafmayer, H. Völzke, M. Seeger, J.F. Miquel, S.C. Sookoian //Gastroenterology. -2010. -Vol. 139, № 6. -P. 1942-1951.
- Gilbert syndrome as a predisposing factor for cholelithiasis risk in the Greek adult population/A. Tsezou, M. Tzetis, E. Giannatou, I. Spanos, E. Roma, A. Fretzayas //Genet. Test. Mol. Biomarkers. -2009. -Vol. 13, № 1. -P. 143-146.
- UGT1A1 promoter polymorphisms and the development of hyperbilirubinemia and gallbladder disease in children with sickle cell anemia/S.L. Carpenter, S. Lieff, T.A. Howard, B. Eggleston, R.E. Ware//Am. J. Hematol. -2008. -Vol. 83, № 10. -P. 800-803.
- Early complication in sickle cell anemia children due to A (TA) nTAA polymorphism at the promoter of UGT1A1 gene/L. Chaouch, E. Talbi, I. Moumni, A.B. Chaabene, M. Kalai, D. Chaouachi //Disease Markers. -2013. -Vol. 35, № 2. -P. 67-72.
- Combined effect of regulatory polymorphisms on transcription of UGT1A1 as a cause of Gilbert syndrome/K. Matsui, Y. Maruo, H. Sato, Y. Takeuchi//BMC Gastroenterology. -2010. -Vol. 10, № 57
- DOI: 10.1186/1471-230X-10-57
- The global distribution of length polymorphisms of the promoters of the glucuronosyltransferase 1 gene (UGT1A1): hematologic and evolutionary implications/A. Premawardhena, C.A. Fisher, Y.T. Liu, I.C. Verma, S. De Silva, M. Arambepola //Blood Cells, Molecules and Diseases. -2003. -Vol. 31. -P. 98-101.
- Genome-wide association meta-analysis for total serum bilirubin levels/A.D. Johnson, M. Kavousi, A.V. Smith, M.H. Chen, A. Dehghan, T. Aspelund //Human Molecular Genetics. -2009. -Vol. 18, № 14. -P. 2700-2710.
- Genome-wide association of serum bilirubin levels in Korean population/T. Kang, H. Kim, H. Ju, J. Kim, Y. Jeon, H. Lee //Human Molecular Genetics. -2010. -Vol. 19, № 18. -P. 3672-3678.
- UDP-Glucuronosyltransferase 1A1 gene polymorphisms and total bilirubin levels in an ethnically diverse cohort of women/A.L. Hong, D. Huo, H.J. Kim, Q. Niu, D.L. Fackenthal, S.A. Cummings //Drug. Metab. Dispos. -2007. -Vol. 35, № 8. -P. 1254-1261.
- Исследование полиморфизмов генов UGT1A1 и DPYD у пациентов с колоректальным раком/Н.Н. Тимошкина, О.А. Богомолова, И.А. Жужеленко, С.Н. Кабанов, Е.А. Калабанова, И.С. Миташок //Сибирский онкологический журнал. -2018. -Т. 17, № 6. -С. 49-56.
- Association of polymorphisms in SULT1A1 and UGT1A1 genes with breast cancer risk and phenotypes in Russian women/E.G. Shatalova, V.I. Loginov, E.A. Braga, T.P. Kazubskaia, M.A. Sudomoina, R.L. Blanchard, O.O. Favorova//Mol. Biol. -2006. -Vol. 40, № 2. -P. 228-234.
- Genotype frequencies of UDP-Glucuronosyltransferase 1A1 promoter gene polymorphism in the population of healthy Croatian pre-scholars/N. Marinkovic, D. Pasalic, B. Grskovic, G. Ferencak, L. Honović, A.S. Rukavina//Coll. Antropol. -2008. -Vol. 32, № 3. -P. 725-729.
- Contribution of the TATA-box genotype (Gilbert syndrome) to serum bilirubin concentrations in the Italian population/M.L. Biondi, O. Turri, D. Dilillo, G. Stival, E. Guagnellini//Clinical Chemistry. -1999. -Vol. 45, № 6. -P. 897-898.
- Polymorphism in the promoter region of the bilirubin UDP-glucuronosyltransferase (Gilbert's syndrome) in healthy Dutch subjects/R.H. Te Morsche, P.L. Zusterzeel, M.T. Raijmakers, E.M. Roes, E.A. Steegers, W.H. Peters//Hepatology. -2001. -Vol. 33, № 3. -P. 765.
- Distribución del genotipo A (TA) 7TAA asociado al. síndrome de Gilbert en la población española/J.M.F. Salazara, Á.R. Sevilla, E. Del Río Conde, M.B. Bastús//Med. Clin. (Barcelona). -2000. -Vol. 115. -P. 540-541.
- UGT1A1 and UGT1A9 functional variants, meat intake, and colon cancer, among Caucasians and African Americans/H. Girard, L.M. Butler, L. Villeneuve, R.C. Millikan, R. Sinha, R.S. Sandler, C. Guillemette//Mutat. Res. -2008. -Vol. 644, № 1-2. -P. 56-63.
- Ботвиньев О.К., Дубровина Г.М., Колотилина А.И. Поражение отделов желудочно-кишечного тракта у детей с синдромом Жильбера//Российский вестник перинатологии и педиатрии. -2015. -№ 3. -С. 104-107.
- Генодиагностика мутаций UGT1A1 в практике современной медицины/А.Н. Волков, С.М. Хабиева, Е.Ю. Смирнова, А.Ю. Ларионов//Клиническая лабораторная диагностика. -2018. -Т. 63, № 3. -С. 186-192.
