Наследственные заболевания в практике врача-патологоанатома

Автор: Герасимов Виктор Николаевич, Яковлева Яна Сергеевна, Уренева Регина Валерьевна
Журнал: Ульяновский медико-биологический журнал @medbio-ulsu
Рубрика: Медико-биологические науки
Статья в выпуске: 3, 2017 года.
Бесплатный доступ
В последние годы отмечаются быстрые темпы развития генетики человека и медицинской генетики: в настоящее время изучены более 4 тыс. наследственных заболеваний. Доля наследственной патологии составляет значительную часть в структуре детской заболеваемости, смертности и инвалидности, но имеет место и в зрелом возрасте. Распространенность наследственных заболеваний во многом зависит от географических параметров. Цель - провести анализ случаев наследственных заболеваний, встретившихся в практической работе врача-патологоанатома за последние 10 лет, по материалам аутопсий Ульяновской областной клинической больницы (ГУЗ УОКБ). Материалы и методы. В представленной работе рассмотрены генетические аспекты, распространенность наследственных заболеваний, макро- и микроскопическая картины проявлений наследственной патологии на примере аутопсийных и биопсийных случаев десятилетнего периода работы патологоанатомического отделения Ульяновской областной клинической больницы. Объектом исследования послужил операционный, биопсийный, секционный материал, забранный у больных и умерших в ГУЗ УОКБ в 2005-2015 гг. После рутинной гистологической проводки изготавливались парафиновые срезы, которые окрашивались гематоксилином и эозином. Проведен морфологический анализ гистологических препаратов и протоколов вскрытий из архивных данных патологоанатомического отделения ГУЗ УОКБ. Результаты. Анализируя частоту встречаемости наследственных заболеваний, можно заключить, что в структуре заболеваемости нашего региона наследственная патология представлена весьма обширным рядом болезней, в т.ч. редкими и атипично текущими хромосомными По-видимому, это обусловлено значительной этнической разнородностью Поволжского региона и общероссийской тенденцией к увеличению наследственной патологии в общей структуре заболеваемости.
Наследственные заболевания, ретинобластома, опухоль вильмса, "тритон"-опухоль, болезнь гоше, синдром альпорта
Короткий адрес: https://sciup.org/14113288
IDR: 14113288 | УДК: 616-006.85; | DOI: 10.23648/UMBJ.2017.27.7084
Hereditary diseases in the practice of a pathologist
Recent years have observed rapid rates of development in human genetics and medical genetics: more than 4,000 hereditary diseases have been studied. Hereditary pathology contributes much to child morbidity, mortality and disability, but it can also be found in adulthood. Geographical parameters greatly influence the prevalence of hereditary diseases. The objective of the study is to analyze hereditary diseases in the practice of a pathologist during the last 10-year period, analyzing the autopsies of Ulyanovsk Regional Clinical Hospital. Materials and Methods. In this study we examined genetic aspects, prevalence of hereditary diseases, macro and microscopic pictures of hereditary disease manifestations. We used ten-year autopsy and biopsy protocols of pathoanatomical department of Ulyanovsk Regional Clinical Hospital. The object of the study was surgical, biopsy, and sectional material taken from patients and persons deceased in Ulyanovsk Regional Clinical Hospital in 2005-2015. After routine histology, paraffin sections stained with hematoxylin and eosin were made. The authors carried out morphological analysis of histologic specimen and autopsy protocols using archival data of Ulyanovsk Regional Clinical Hospital pathoanatomical department. Results. Analyzing the incidence of hereditary diseases in the regional morbidity, we conclude that hereditary pathology is represented by a very wide range of diseases, including rare and atypical chromosome abnormalities. It is apparently caused by considerable ethnic heterogeneity of the Volga region; how-ever,hereditary pathology increases in the national morbidity structure.
Текст научной статьи Наследственные заболевания в практике врача-патологоанатома
Введение. Наследственные заболевания - это болезни, появление и развитие которых связано со сложными нарушениями в наследственном аппарате репродуктивных клеток. Возникновение таких недугов обусловлено нарушениями в процессах хранения, реализации и передачи генетической информации. Нередко термины «наследственная болезнь» и «врожденная болезнь» ошибочно употребляются как синонимы, однако врожденными болезнями называют те заболевания, которые имеются уже при рождении ребенка и могут быть обусловлены как наследственными, так и экзогенными факторами. Однако далеко не все наследственные болезни относят к врожденным, поскольку многие из них проявляются только после периода новорожденности [1].
Наследственная патология составляет значительную часть в структуре детской заболеваемости, смертности и инвалидности, но имеет место и в зрелом возрасте [2]. Распространенность наследственных заболеваний во многом зависит от географических параметров. Так, согласно исследованиям А.Р. Магжановой наследственных мутаций населения Волго-Уральского региона, неоднородность этнического состава определяет и частоту встречаемости наследственной патологии. На территории Волго-Уральского региона проживают представители тюркской ветви алтайской языковой семьи (башкиры, татары, чуваши), финно-угорской ветви уральской языковой семьи (марийцы, мордва, удмурты, коми) и славянской ветви индоевропейской языковой семьи (русские). Поэтому здесь встречаются мутации различного происхождения, а их частота и спектр у больных разных этнических групп довольно своеобразны и отличаются от других регионов России [3, 4]. По стране в целом в 2015 г. количество новорожденных с наследственной патологией зарегистрировано: с хромосомными болезнями – 5–8 на 1000 новорожденных, с моноген-ными заболеваниями – 7–17 на 1000 [5].
Цель исследования. Провести анализ случаев наследственных заболеваний, встретившихся в практической работе врача-патологоанатома за последние 10 лет, по материалам аутопсий Ульяновской областной клинической больницы (ГУЗ УОКБ).
Материалы и методы. Объектом исследования послужил операционный, биопсийный, секционный материал, забранный у больных и умерших в ГУЗ УОКБ в 2005– 2015 гг. После рутинной гистологической проводки изготавливались парафиновые срезы, которые окрашивались гематоксилином и эозином. Проведен морфологический анализ гистологических препаратов и протоколов вскрытий из архивных данных патологоанатомического отделения ГУЗ УОКБ. При морфоскопии использованы исследовательский бинокулярный микроскоп Carl Zeiss, увеличение 100 и 400, и цифровая камера MMC-31M-12C.
Результаты и обсуждение. Одним из тяжелых наследственных заболеваний является опухоль глаза – ретинобластома. Чаще всего ретинобластома обусловлена генетически. Как правило, речь идет о делеции участка длинного плеча одной из хромосом 13-й пары с повреждением гена Rb1. Случаи, когда у родителей, болевших ретинобластомой, рождаются здоровые дети, встречаются довольно редко [6].
Пример из практики. У мужчины 67 лет проведена энуклеация глаза, материал прислан на исследование с диагнозом «терминальная болящая глаукома». Макроскопически: глазное яблоко 4,0×4,5 см, роговица тусклая, в области сетчатки образование 1,0×0,6 см, на разрезе ткань бледно-серого цвета с кровоизлияниями. При микроскопии в сетчатке глаза наблюдался рост недифференцированной ретинобластомы, которая представлена мелкими мономорфными клетками с крупным ядром (рис. 1).
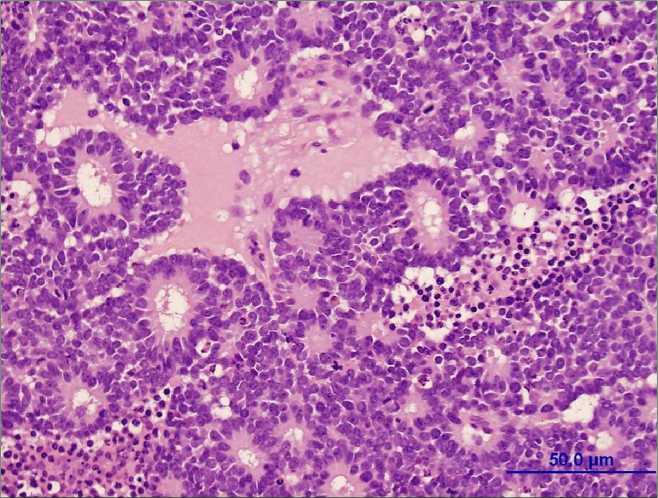
Рис. 1. Ретинобластома.
Окраска гематоксилином и эозином, ув. ×40
Ретинобластома – это опухоль детского возраста: на ее долю приходится 2,5–4,5 % случаев всех злокачественных новообразований у детей до 15 лет. В большинстве случаев ретинобластома развивается в возрасте до 5 лет, пик заболеваемости приходится на 2–3 года [7]. В приведенном выше случае опухоль глаза диагностирована у мужчины 67 лет, что свидетельствует о нетипичном развитии данной патологии.
Другой опухолью, передающейся по наследству, является опухоль Вильмса. Опухоль Вильмса (ОВ) составляет примерно 20–25 % всех злокачественных новообразований у детей, её обнаруживают у детей разного возраста, однако чаще – от 2 до 5 лет. Очень редко опухоль Вильмса встречается у взрослых: только у 0,9 % всех пациентов с опухолью почки. Установлено, что ОВ связана с нарушением эмбриогенеза почки и ключевая роль отведена генетическим нарушениям. За развитие ОВ ответственны нарушения в нескольких генах: WT1, WT2 и WT3. Первым из них был идентифицирован в 1989 г. ген WT1. Он локализован на коротком плече 11-й хромосомы и отвечает за образование специфических белков, участвующих в регу- ляции развития первичного нефрона. В норме продукты гена WT1 выступают в качестве регулятора развития почки и супрессора опухолевого роста. Помимо гена WT1, на хромосоме 11р идентифицирован ген WT2, а на хромосоме 16q – ген WT3. Предполагают нарушения 1р, 7р, 17р, 19q. Кроме выявленных нарушений в определенных генах, в последние годы важная роль в развитии ОВ отводится дисрегуляции фетальных митогенов – повышению экспрессии гена инсулиноподобного фактора роста [8] .
Пример из практики. У женщины 39 лет была удалена почка с опухолевидным образованием. Макроскопически: почка в капсуле размером 8,5×6,5 см. Капсула тонкая, снимается легко. Поверхность почки крупнобугристая. У наружного края почки под капсулой обнаружен узел размером 3×4 см, на разрезе – бледно-серого цвета с наличием множества мелких кист. На отдельных участках узел связан с чашечками и лоханками. Паренхима почки красно-коричневая, границы слоев различимы. Микроскопическая картина соответствовала мезенхимальной нефробла-стоме (опухоль Вильмса) (рис. 2).
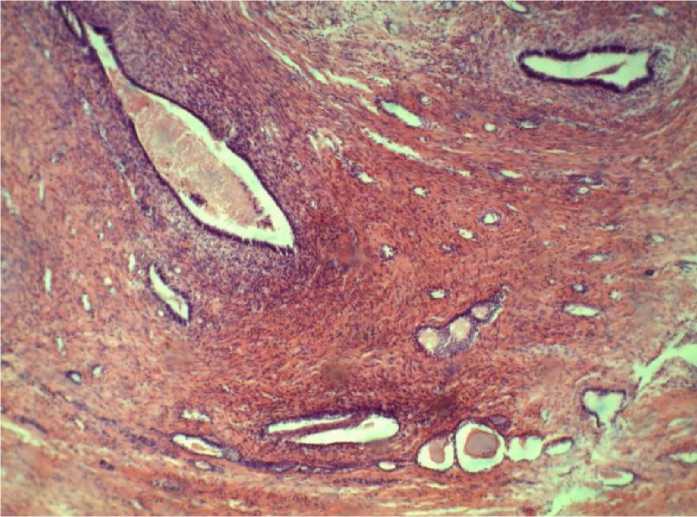
Рис. 2. Мезенхимальная нефробластома (опухоль Вильмса). Окраска гематоксилином и эозином, ув. ×40
Вопреки тому, что опухоль Вильмса развивается преимущественно у маленьких детей, в медицинской практике встречаются единичные случаи этого заболевания у пациентов, возраст которых вписывается в диапазон от 6 до 65 лет [9]. Учитывая крайнюю редкость этой патологии у взрослых пациентов, необходимо отметить отсутствие систематизированных сведений об особенностях ее клинического течения, лечения и прогнозе у данной категории больных.
Крайне редко встречающейся наследственной опухолью является «тритон»-опухоль. Это злокачественная опухоль оболочек, на долю которой приходится до 10 % всех мягкотканных сарком. Она может развиваться de novo или путем озлокачествления таких опухолей, как шваннома и ганглионеврома, нередко ассоциируется с нейрофиброматозом Реклингхаузена. Опухоль имеет дифферен- цировку рабдомиосаркомы и может включать в себя участки хондросаркомы, остеосаркомы, а также эпителиальные железистые структуры [10].
Пример из практики. У мужчины 45 лет обнаружена опухоль правой теменной доли, которая удалена оперативным путем. Макроскопическая картина: узел неправильной формы 3×3 см с бугристой поверхностью, белесовато-желтого цвета. На разрезе ткань плотная, бледно-серая, слоистая с участками бледно-желтого цвета. При микроскопическом исследовании можно было предположить метастазирование остеогенной саркомы в ткань головного мозга. После проведения консилиума выставлен диагноз «опухоль мозга состоит из участков типа рабдомиосаркомы и остеогенной саркомы, что может иметь место при «тритон»-опухоли» (рис. 3).
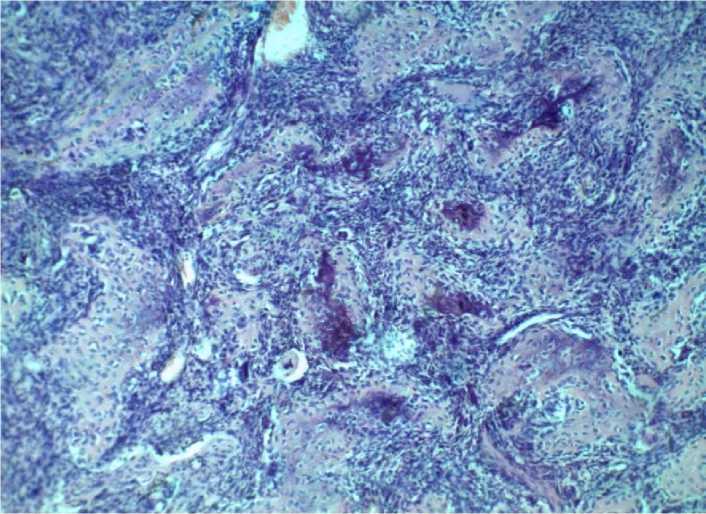
Рис. 3. «Тритон»-опухоль. Окраска гематоксилином и эозином, ув. х40
«Тритон»-опухоль обладает высоким злокачественным потенциалом и представляет собой классический пример морфологически сложного образования, сочетающего в себе два начала. Биологический феномен данной опухоли является в какой-то мере моделью для изучения подобных опухолей. Данные обстоятельства заставляют по-осо- бому взглянуть на это заболевание и являются важной посылкой к их рассмотрению.
Кроме опухолей с наследственной предрасположенностью, в 10-летней практике нашего отделения встречались и случаи наследственных синдромов, одним из которых является нарушение обмена веществ в клетке - болезнь Гоше. Заболевание описал в
1882 г. француз Филипп Шарль Эрнст Гоше. В основе болезни Гоше лежит наследственный дефицит активности фермента (глюко-цереброзидазы), участвующего в переработке продуктов в-фермента клеточного метаболизма. В результате недостаточной активности этого фермента в макрофагах накапливаются непереработанные «отходы» метаболизма жиров, которые располагаются сначала в селезенке, затем в печени, костях, костном мозге, легких. Болезнь Гоше встречается с частотой от 1:60 000 до 1:40 000 у представителей всех этносов; в популяции евреев Ашкенази частота достигает 1:450 [11].
Пример из практики. У мужчины 49 лет клинически диагностирован цирроз печени с гепатоспленомегалией. Оперативным путем удалена селезенка, взят участок печени на гистологическое исследование. Макроскопическая картина: селезенка 17,0×11,5 см, капсула тонкая. На разрезе паренхима селезенки мягко-эластичная, бледно-вишневая, однородная. При поскабливании обушком ножа соскоба не дает. Кусочек печени 0,5×0,4 см, паренхима бледно-коричневого цвета. При исследовании гистологических препаратов селезенки в красной пульпе имеются инфильтраты и диффузные скопления крупных светлых клеток с бледно-розовой цитоплазмой и эксцентрически расположенным ядром типа клеток Гоше. Лимфоидные фолликулы сохранены, синусы расширены. В паренхиме печени - очаговая жировая дистрофия гепатоцитов без признаков цирроза. Описанная морфологическая картина соответствует диагнозу «сфинголипидоз (болезнь Гоше)» (рис. 4). Болезнь Гоше можно диагностировать с помощью молекулярного анализа гена глюкоцереброзидазы [12]. Однако такой метод крайне сложный и дорогостоящий, поэтому к нему прибегают в редких случаях, когда диагностика заболевания затруднена. Наиболее часто диагноз ставят при обнаружении клеток Гоше в пункции костного мозга или увеличенной селезенки при проведении биопсии.
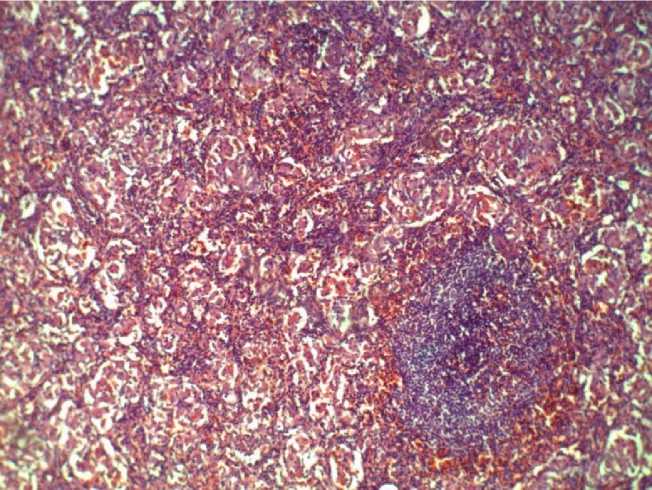
Рис. 4. Селезенка при болезни Гоше. Окраска гематоксилином и эозином, ув. х40
Другим примером наследственного синдрома является синдром Альпорта. Синдром Альпорта (СА) представляет собой заболевание, вызываемое мутациями в генах, кодирующих различные а-субъединицы коллагена IV типа. Причиной заболевания служит му- тация одного из генов: COL4A5, COL4A4, COL4A3. При классическом варианте СА мутация происходит в гене COL4A5, расположенном на длинном плече Х-хромосомы (Xq22.2), который кодирует а5-цепь коллагена IV типа. Аутосомно-рецессивная форма синдрома Альпорта обусловлена мутацией генов CОL4A3 и COL4A4, расположенных на хромосоме 2 и кодирующих соответственно а3- и а4-цепи коллагена этого типа. Аутосомно-доминантная форма СА сцеплена с генным локусом COL4A3–COL4A4. Распространенность заболевания составляет 1 на 5000 случаев [13].
Пример из практики . Умерший больной М., 19 лет. С возраста 10 лет состоял на учете в детской больнице с диагнозом «наследственный нефрит; тугоухость». Из анамнеза: у дяди больного был синдром Альпорта. Больной неоднократно лечился стационарно, в последнюю госпитализацию уровень мочевины составлял 44,6 ммоль/л, креатинина –
1678 мкмоль/л. В рамках лечения проводились сеансы гемодиализа. По результатам вскрытия – почки гипоплазированны. Общая масса почек 100 г, левая – 40 г, правая – 60 г, капсула снялась с трудом, поверхность почек мелкозернистая. На разрезе паренхима анемичная, пирамидки темно-синюшные, корковый слой истончен, границы между слоями четкие. Лоханки не расширены, мочеточники диаметром 0,4 см, проходимы. При микроскопическом исследовании почек – клубочки с утолщенной фиброзной капсулой, в некоторых капсула заращена, с очагами лимфоги-стиоцитарной инфильтрации, переходящей в очаговый фиброз и атрофию. Сосуды – с фиброзом стенки и ее гомогенизацией (рис. 5).
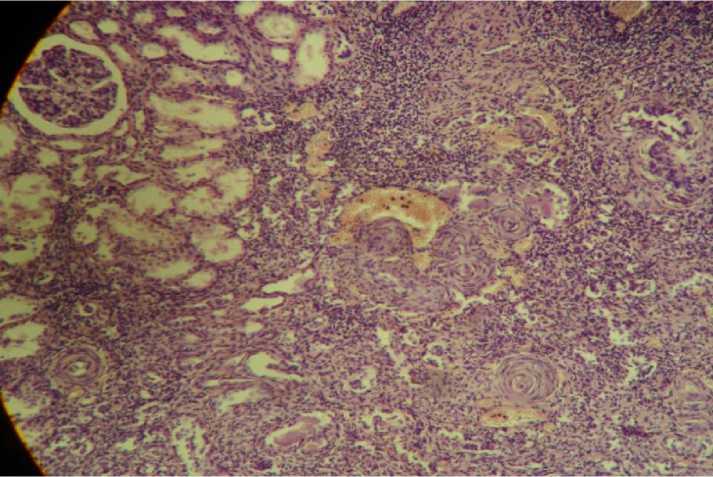
Рис. 5. Почки при синдроме Альпорта. Окраска гематоксилином и эозином, ув. ×40
У лиц мужского пола с Х-сцепленным доминантным вариантом СА заболевание имеет прогрессирующее течение: терминальная ХПН развивается у 50 % больных в возрасте до 25 лет [14]. При делеции или нонсенс-мутации COL4A5 риск развития терминальной ХПН в возрасте до 30 лет составляет 90 %. В большинстве случаев сроки развития терминальной ХПН у мальчиков с Х-сцеп-ленным доминантным вариантом СА схожи с другими членами семьи мужского пола, что позволяет прогнозировать время наступления терминальной ХПН даже без определения ге- нотипа, что и наблюдалось в приведенном выше случае [15].
Заключение. В последние годы отмечаются быстрые темпы развития генетики человека и медицинской генетики: в настоящее время изучены более 4 тыс. наследственных заболеваний. Это объясняется многими причинами и прежде всего резким увеличении-ем доли наследственной патологии в структуре заболеваемости и смертности населения. Анализируя частоту встречаемости наследственных заболеваний по результатам 10-летней практики патологоанатомичес- кого отделения Ульяновской областной клинической больницы, можно заключить, что в структуре заболеваемости нашего региона наследственная патология представлена весьма обширным рядом заболеваний, в т.ч. редкими и атипично текущими хромосомны- ми болезнями. По-видимому, это обусловлено значительной этнической разнородностью Поволжского региона и общероссийской тенденцией к увеличению наследственной патологии в общей структуре заболеваемости.
Список литературы Наследственные заболевания в практике врача-патологоанатома
- Бочкова Н.П. Медицинская генетика. М.: Мастерство; 2002. 656.
- Иванов В.И. Генетика: учебник для вузов. М.: Академкнига; 2006: 359-360.
- Кириллов А.Г. Наследственные болезни в Чувашской Республике: автореф. дис.. д-ра мед. наук. М.; 2008. 52.
- Кулешов Н.П., Мутовин Г.Р., Марченко Л.Ф. Организационные и методические основы пренатальной диагностики в профилактике наследственных и врожденных болезней. М.; 2004. 112.
- Потапова О.Н. Медико-социологическая характеристика феномена детской инвалидности. Социология медицины. 2010; 1: 11-17.
- Мякова Н.С. Ретинобластома. В кн.: Румянцев А.Г., Самочатова Е.В., ред. Гематология, онкология детского возраста. М.: Медпрактика; 2004. 679-684.
- Аксель Е.М., Горбачева И.А. Заболеваемость детей злокачественными новообразованиями и смертность от них в России и странах СНГ. Вестник РОНЦ им. Н.Н. Блохина РАМН. 2007; 2: 136-154.
- Дурнов Л.А. Нефробластома. Опухоль Вильмса в свете проблем детской онкологии. Российский вестник перинатологии и педиатрии.1995; 4: 10-13.
- Аксель Е.М., Двойрин В.В., Дурнов Л.А. Статистика злокачественных новообразований детей в России. Вопросы онкологии. 1997; 4: 371-384.
- Петровичев Н.Н., Еремина Л.А., Хмелев О.Н., Касумов Н.В. Низкодифференцированная (дедиф-ференцированная) хондросаркома. Вопросы онкологии. 1984; 1: 8-13.
- Краснопольская К.Д. Наследственные болезни обмена веществ. М.: Центр социальной адаптации и реабилитации детей «Фохат»; 2005. 364.
- Sardi S.P., Viel C., Clarke J., Treleaven C.M. Glucosylceramide synthase inhibition alleviates aberrations in synucleinopathy models. Proc. Natl. Acad. Sci. USA; 2017. pii: 201616152.
- Игнатова М.С. Детская нефрология: руководство для врачей. М.: ООО «Медицинское информационное агентство»; 2011. 696.
- Phelan P.J., Fletcher E., Carroll N., Metcalfe W., Turner A.N. Simultaneous adult polycystic kidney disease and Alport syndrome. Nephrology (Carlton). 2016; 8: 722-723.
- Вельтищев Ю.Е., Игнатова М.С. Профилактическая и превентивная нефрология. М.; 1996. 61.
