Немалиновая миопатия редкой формы (случай из практики)
")
Автор: Мигалкин Николай Сергеевич, Филимонова Галина Николаевна, Прудникова Оксана Германовна, Мезенцев Игорь Николаевич
Журнал: Гений ортопедии @geniy-ortopedii
Рубрика: Случай из практики
Статья в выпуске: 5 т.28, 2022 года.
Бесплатный доступ
Введение. Немалиновые миопатии - группа нейромышечных заболеваний, отличительной гистологической особенностью которых является наличие немалиновых тел в миосимпластах. Цель. Описательная морфология редкой формы первичной немалиновой миопатии, прогрессирующей в позднем возрасте. Материалы и методы. Исследован операционный материал паравертебральных мышц пациентки 51 года со сколиотической деформацией на уровне L4-S1, оперированной неоднократно с целью коррекции деформации позвоночника на фоне неврологических нарушений. Парафиновые срезы окрашивали гематоксилином и эозином, по Массону, методом Ptah, изучали посредством сканирующего микроскопа 3DHISTECH Pannoramic MIDI II BF для оцифровки препаратов по технологии Whole slide imaging в двух режимах: Single layer mode и Extended focus (3DHISTECH, Венгрия). Результаты. Во фрагментах измененной мышечной ткани в миосимпластах идентифицировались нитчатые структуры немалиновых тел, которые располагались диффузно-точечно по всей саркоплазме либо образовывали скопления различной конфигурации. Наблюдалась повышенная вариабельность диаметров мышечных волокон, внутренние ядра, контрактурно измененные миосимпласты и с признаками миофагий, картины постепенного замещения мышечных волокон адипоцитами, массовая жировая дегенерация волокон, фиброз интерстициального пространства. Внутримышечные нервные стволики имели признаки полной инволюции, сохранялся фиброзированный периневрий, единичные нервные волокна; нервно-мышечные веретена отличались увеличенной соединительнотканной капсулой. Сосуды артериального звена с признаками фиброза и облитерацией просвета, сосуды венозного русла извитые. Обсуждение. В связи с большим числом генов, ответственных за NM, генетический поиск может быть затруднен и бывает результативным лишь в 50 % случаев. Установлено, что немалиновые тела могут распределяться диффузно либо образовывать скопления неправильной формы, чаще субсарколеммально и характерны для мелких волокон. В представленном клиническом случае немалиновые тела наблюдались по всей площади волокон и были характерны для миосимпластов различных размеров. Заключение. Посредством патогистологического исследования паравертебральных мышц была установлена нейромышечная природа заболевания, определенного как немалиновая миопатия, прогрессирующая в позднем возрасте, не диагностированная ранее на предыдущих этапах лечения.
Немалиновая миопатия, паравертебральные мышцы, немалиновые образования, фиброз, жировая дегенерация, инволюция нервных проводников, облитерация сосудов
Короткий адрес: https://sciup.org/142236796
IDR: 142236796 | УДК: 616.741-009 | DOI: 10.18019/1028-4427-2022-28-5-715-719
Rare nemaline myopathy (a case report)
Introduction Nemaline myopathies (NM) are a group of neuromuscular diseases, the distinctive histological feature of which are nemaline rods in myosymplasts. The purpose of this work is to describe the morphology of a rare form of primary nemaline myopathy that progresses in adulthood. Material and methods The surgical material of the paravertebral muscles of a 51-year-old patient with scoliotic deformity at the level of L4-S1, who was repeatedly operated on to correct spinal deformity due to neurological disorders, was studied. Paraffin sections were stained with hematoxylin-eosin, according to Masson, using the Ptah method, studied using a 3DHISTECH Pannoramic MIDI II BF scanning microscope to digitize preparations using Whole slide imaging technology in two modes: Single layer mode and Extended focus (3DHISTECH, Hungary). Results In the fragments of the altered muscle tissue, filamentous structures of nemaline bodies in myosymplasts were identified, which were located diffusely-dotted throughout the sarcoplasm or formed clusters of various configurations. There was an increased variability in the diameters of muscle fibers, internal nuclei, myosymplasts altered by contraction and with signs of myophagy, patterns of gradual replacement of muscle fibers by adipocytes, massive fatty degeneration of fibers, and fibrosis of the interstitial space. Intramuscular nerve trunks showed signs of complete involution; fibrous perineurium was preserved, and there were single nerve fibers; neuromuscular spindles were distinguished by an enlarged connective tissue capsule. The vessels of the arterial flow had signs of fibrosis and obliteration of the lumen; the vessels of the venous bed were tortuous. Discussion Due to a large number of genes responsible for NM, genetic search can be difficult and is effective only in 50 % of cases. It has been established that nemaline bodies can be distributed diffusely or form clusters of irregular shape, more often subsarcolemmal and characteristic of small fibers. In the presented clinical case, nemaline bodies were observed over the entire area of the fibers and were characteristic of myosymplasts of various sizes. Conclusion The histopathological study of the paravertebral muscles established the neuromuscular nature of the disease, being nemaline myopathy that progressed in adulthood and had not been diagnosed at previous stages of treatment.
Текст научной статьи Немалиновая миопатия редкой формы (случай из практики)
Немалиновые миопатии (Nemaline myopathy, NM) – группа прогрессирующих нейромышечных заболеваний, которые характеризуются наличием аномальных скрученных или палочковидных структур в мышеч- ных волокнах, выявляемых при гистопатологическом исследовании (NEMA – «нить»). Болезнь относится к наиболее распространенным миопатиям, частота встречаемости один на 50000 человек в равной степени
мужчины и женщины, обычно передается по наследству, в единичных случаях заболевание не наблюдается у других членов семьи [1, 2]. Клинические проявления NМ значительно варьируют как по возрасту манифестации, так и тяжести течения, в 90 % случаев миопатии проявляются при рождении или в раннем детстве, очень редко обнаруживаются у взрослых [3]. Отличительными чертами NМ являются гипотония, слабость скелетных мышц, деформации позвоночника, проблемы при кормлении [4].
Выделяют шесть клинических групп NM в зависимости от возраста манифестации и степени тяжести. Взаимосвязь между тяжестью заболевания и определенным геном, в котором произошла мутация, не установлена, необходимо регулярное наблюдение у кардиолога [4]: 1) тяжелая врожденная форма: сильная мышечная гипотония, нарушение функции внешнего дыхания; 2) немалиновая миопатия Амишей при рождении: гипотония мышц, контрактуры суставов, нарушения дыхания; 3) промежуточная врожденная форма: средняя степень тяжести, задержка моторного развития, утрата способности самостоятельной ходьбы; 4) типичная (мягкая) врожденная форма: гипотония, вялость в мышцах, расположенных ближе к туловищу; 5) миопатия, проявляющаяся в 8-15 лет: нормальное раннее моторное развитие, симметричная слабость мышц лодыжек; 6) миопатия, проявляющаяся во взрослом возрасте 20-50 лет: быстро прогрессирующая общая слабость мышц, боль в мышцах, деформации позвоночника. Механизмы, приводящие к формированию немалиновых миопатий, являются предметом продолжающихся исследований [1, 3, 5, 6].
Представляемый клинический случай интересен не только с точки зрения особенностей клинической картины заболевания (стертость манифестации неврологических проявлений), но и особенностей гистоструктуры мышечной ткани при патогистологическом анализе. В рамках клинической работы представлена краткая морфологическая характеристика и обсуждение данной проблемы [7].
Цель работы – описательная морфология редкой формы немалиновой миопатии, прогрессирующей в позднем возрасте.
МАТЕРИАЛЫ И МЕТОДЫ
Исследован операционный материал параспиналь-ных мышц пациентки 51 года со сколиотической деформацией позвоночника на уровне L4-S1. При поступлении была диагностирована неврологическая симптоматика в виде болезненных спазмов мышц лица, шеи, гипотонуса и гипотрофии мышц бедер, голеней, стоп, ограничений движений в конечностях. Прогрессирующий неврологический дефицит не укладывался в клинические проявления вторичной миопатии, после сбора анамнеза было принято решение о проведении гистопатологического исследования мышц спины с целью уточнения этиологии заболевания.
Мышечную ткань фиксировали в 10 % растворе нейтрального формалина, после гистологической про- водки заливали в парафин, срезы окрашивали гематоксилином и эозином, трихромным методом по Массону, методом PTAH. Изучали посредством сканирующего микроскопа 3DHISTECH Pannoramic MIDI II BF для получения цифровых микропрепаратов по технологии Whole slide imaging в двух режимах: Single layer mode и Extended focus (3DHISTECH, Венгрия).
Обследования пациентки проводились в соответствии с требованиями Хельсинской декларации 1975 года, пересмотренными в 2013 году; было подписано информированное добровольное согласие на осуществление диагностики и медицинского вмешательства с дальнейшим использованием данных в научных целях.
РЕЗУЛЬТАТЫ
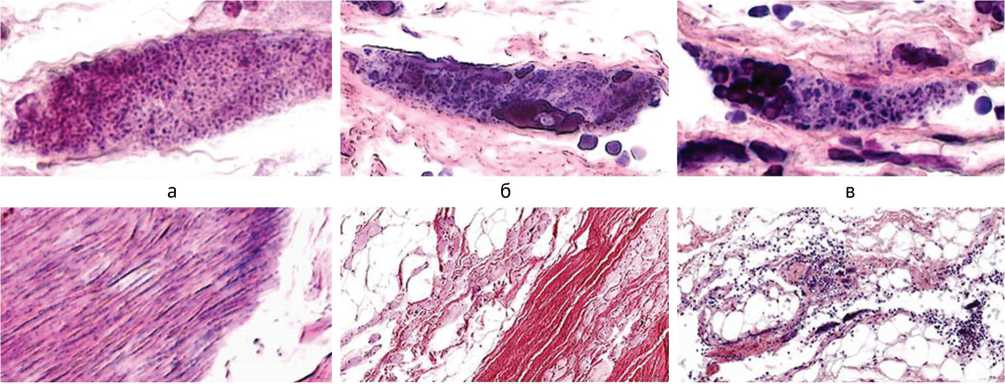
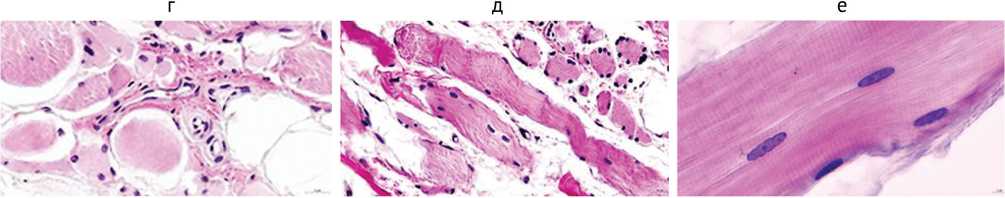
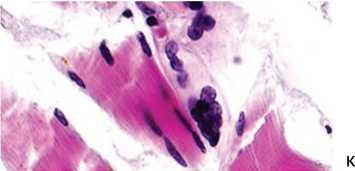
Проведенное патогистологическое исследование паравертебральных мышц пациентки, неоднократно оперированной с целью коррекции деформации позвоночника на фоне неврологических нарушений, позволило уточнить этиологию основного заболевания и дифференцировать его как генетически обусловленную немалиновую миопатию, прогрессирующую в позднем возрасте. Макроскопически наблюдались фрагменты мышечной ткани с выраженным фиброзом, атрофией, жировым замещением. При микроскопировании измененной мышечной ткани визуализировались миосимпласты, под сарколеммой которых идентифицировались немалиновые тела, окрашенные темно-синим по Рtah (рис. 1). Одни располагались диффузно в виде точечных вкраплений (рис. 1, а), другие группировались в кластеры разнообразной конфигурации (рис. 1, б, в). На продольных срезах немалиновые образования визуализировались в виде нитчатых структур (рис. 1, г). В операционном материале наблюдались поля адипоцитов, заместивших мышечные волокна, различные этапы преобразования миосимпластов в жировые клетки, остаточные мелкие миоциты погружа- лись в соединительнотканные конгломераты (рис. 1, д), нередко встречались геморрагии (рис. 1, е). Были характерны волокна разнообразных диаметров, визуализировались картины постепенного замещения миосимпластов жировыми клетками (рис. 1, ж), отмечались внутренние ядра и контрактурно измененные волокна (рис. 1, з). В симпластах с различимой поперечной исчерченностью идентифицировались вытянутые собственные ядра по периферии и в центре (рис. 1, и), наряду с сохранными фрагментами мышечной ткани наблюдались мышечные волокна с утраченной гистоструктурой (рис. 1, к).
Сосуды зачастую были погружены в конгломераты адипоцитов, просветы облитерированы, оболочки с признаками фиброза, гладкомышечные клетки (ГМК) t. media с нарушенной циркулярной ориентацией (рис. 2, а, б). Сосуды венозного русла тонкостенные, извитые (рис. 2, в). Нервно-мышечные веретена характеризовались увеличенной соединительнотканной капсулой (рис. 2, г), внутримышечные нервные проводники подвергались полной инволюции, сохранялся фиброзированный периневрий, единичные нервные волокна (рис. 2, д).
3 и
Рис. 1. Фрагменты парафиновых срезов параспинальных мышц при немалиновой миопатии: а – миоцит с диффузным распределением немалиновых стержней; б, в – гонгломераты немалиновых тел; в – слева группа клеток макрофагального ряда в миосимпласте с немалиновыми телами; г – темно-синие немалиновые нити; д – остаточные мышечные волокна погружены в поле адипоцитов, справа – волокна, заключенные в соединительнотканное разрастание; е – геморрагии; ж – различные этапы замещения мышечных волокон на адипоциты, волокна разнообразных диаметров; з – контрактурно измененные миосимпласты, внутренние ядра; и – сохранные мышечные волокна; к – миосимпласт с утраченной гистоструктурой (справа). Окраска: а–е – методом Pyah; ж–к – гематоксилином и эозином. Увеличение: а, б, в – 2300×; г – 3000×; д – 200×; е – 300×; ж – 700×; з – 500×; и – 1100×; к – 1600×
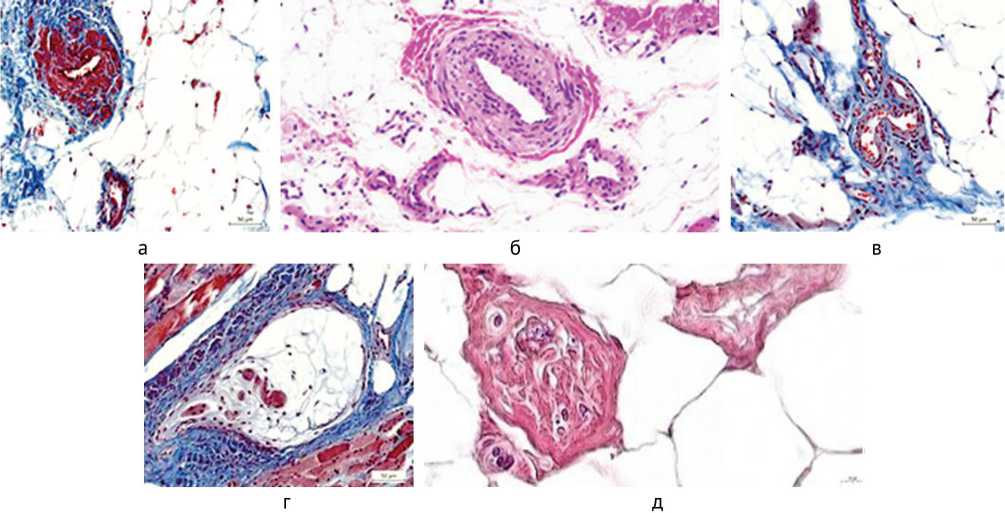
Рис. 2. Фрагменты парафиновых срезов паравертебральных мышц при немалиновой миопатии, особенности гистоструктуры сосудов, нервно-мышечных веретен и нервных проводников: а – сосуды погружены в скопления адипоцитов, окклюзия просвета; б – выраженный фиброз t. media, ГМК лишены циркулярной ориентации; в – извитые контуры венозного сосуда; г – увеличенная капсула нервно-мышечного веретена в соединительнотканном конгломерате; д – инволюция внутримышечного нервного стволика с массовой дегенерацией нервных волокон, сохранен фиброзированный периневрий. Окраска: а, в, г – по Массону, б – гематоксилином и эозином, д – методом Ptah. Увеличение: а, в, г – 500×; б – 270×; д – 1600×
ОБСУЖДЕНИЕ
В исследованном клиническом случае имеет место патология, проявившаяся в подростковом возрасте и неуклонно прогрессирующая во взрослом. По данным литературы, в связи с большим числом генов, ответственных за NM, генетический поиск может быть затруднен и бывает результативным лишь в 50 % случаев [1], необходимы дополнительные исследования, в частности, патогистологический анализ.
Фиброз стенок сосудов, обусловленный чрезмерным отложением белков внеклеточного матрикса, связывают с такими заболеваниями как атеросклероз, гипергликемия, дислипидемия, гипергомоцистеинемия; а также с ренин-ангиотензин-альдостероновой системой, окислительным стрессом, факторами роста и дисбалансом секреции цитокинов, полученных из эндотелия [8]. Дисбаланс коллагенов приводит к фиброзу стенки и, как следствие, к облитерации просвета; установлена роль коллагенов в обеспечении гемостаза, с акцентом на коллагены типов I–IV, VI, XV, XVIII [9]. Показана также роль ГМК в развитии жесткости стенок артерий, это второй важный компонент после белков внеклек-точного матрикса, который не только регулирует взаимодействия актомиозина при сокращении, но и опосредует механотрансдукцию в клетках и гемостаз белков внеклеточного матрикса. Жесткость артерий, обратная растяжимости, вовлечена в этиологию хронических сердечнососудистых заболеваний и является основной причиной заболеваемости и смертности во всем мире [10].
Данные, представленные в настоящей работе, находят отражение в других исследованиях. Известно, особенностями мышечной ткани при NM являются аномальная вариабельность размеров мышечных волокон, что обусловливает бимодальное распределение диметров на гистограмме; внутренние ядра, локальный некроз мышечных волокон, выраженный фиброз эндо- и перимизия [11]. Также установлено, что немалиновые тела могут распределяться диффузно либо образовывать скопления неправильной формы, чаще субсар-колеммально, и характерны для мелких волокон [12]. Последние 2 факта не подтверждены в представленной работе, немалиновые образования наблюдались не только под сарколеммой, но по всей площади волокон, и в равной степени были характерны для миосимпла-стов различных размеров.
Методом электронной микроскопии выявлено, что идентифицировать NM можно по наличию структур стержневидной или яйцевидной формы; немалиновые стержни имеют аналогичную Z-дискам решетчатую структуру, их форма у пациентов с мутациями в KLHL40 и LMOD3 различна и может быть полезна для диагностики [4].
Установлено, что нарушения сократительной способности и организации цитоскелета при NM вызывают такие дефекты ядра как разрушенная оболочка, измененное расположение хроматина, что, в свою очередь, учитывая важную роль ядра в регуляции экспрессии генов, а также цитоскелета в поддержании целостности миосимпластов, объясняет нарушение сократительной способности филаментов [12].
Тонкая нить актина – основная составляющая саркомера, именно на этом уровне сокращения возникает мышечная слабость у пациентов с NМ [3]. Десять из 12 известных генов при NM кодируют белки, которые являются компонентами тонкого филамента, либо способствуют стабильности и обновлению актина. Белки, кодирующиеся генами NEM2 и ACTA1 , участвуют в формировании мышечного тонуса и сокращении мышц. NEM2 – кодирует белок nebulin, мутации здесь являются основной причиной возникновения миопатии и обнаружены в 66,7 % случаев [5]. Показано как мыши с дефицитом nebulinа демонстрировали тяжелую мышечную атрофию и слабость in vivo, связанные с низким содержанием NEМ2 [13]. Посредством морфологического анализа мышц с мутацией в NEМ2 установлено, что количество волокон, занятых немалиновыми телами, обратно коррелируют с тяжестью болезни [14]. Аномальная укладка, измененная полимеризация и агрегация мутантных изоформ α -actinа являются общими свойствами мутантов при NM, некоторые из этих эффектов специфичны и, вероятно, приводят к вариациям в степени мышечной слабости [6].
Понимание вклада саркомерной дисфункции в мышечную слабость через задействованные гены поможет разработать целевые терапевтические стратегии [3]. Точная диагностика заболевания позволит определять тактику лечения и избегать неблагоприятных исходов применяемых хирургических методов [15, 16].
ЗАКЛЮЧЕНИЕ
Посредством патоморфологического исследования паравертебральных мышц была установлена нейромышечная природа заболевания, определенного как немалиновая миопатия, прогрессирующая в позднем возрасте, не диагностированная ранее на предыдущих этапах лечения. Для мышц характерны немалиновые тела двух типов, локализующиеся диффузно-точечно в саркоплазме либо образующие конгломераты различной формы, как под сарколеммой, так и по всей площади волокон; повышенная вариабельность диаметров миосимпластов, внутренние ядра, контрактурно из-
мененные волокна, картины миофагий. Наблюдались поля жировой дистрофии, инволюция нервных проводников с фиброзированным периневрием, фиброз интерстициального пространства и стенок сосудов, их облитерация.
При сложных деформациях позвоночника необходима оценка неврологического статуса и сбор анамнеза с использованием максимального спектра тестов, патогистологический анализ операционного материала является одним их объективных диагностических критериев.
Список литературы Немалиновая миопатия редкой формы (случай из практики)
- Malfatti E., Romero N.B. Nemaline myopathies: State of the art // Rev. Neurol. (Paris). 2016. Vol. 172, No 10. P. 614-619. DOI: 10.1016/j. neurol.2016.08.004.
- Nemaline myopathies / C. Wallgren-Pettersson, C.A. Sewry, K.J. Nowak, N.G. Laing // Semin. Pediatr. Neurol. 2011. Vol. 18, No 4. P. 230-238. DOI: 10.1016/j.spen.2011.10.004.
- De Winter J.M., Ottenheijm C.A.C. Sarcomere. Dysfunction in Nemaline Myopathy // J. Neuromuscul. Dis. 2017. Vol. 4, No 2. P. 99-113. DOI: 10.3233/JND-160200.
- Sewry С.А., Laitila J.M., Wallgren-Pettersson C. Nemaline myopathies: a current view // J. Muscle Res. Cell Motil. 2019. Vol. 40, No 2. P. 111-126. DOI: 0.1007/s10974-019-09519-9.
- Clinico-pathological features and mutational spectrum of 16 nemaline myopathy patients from a Chinese neuromuscular center / X. Yin, C. Pu, Z. Wang, K. Li, H. Wang // Acta Neurol. Belg. 2022. Vol. 122, No 3. P. 631-639. DOI: 10.1007/s13760-020-01542-9.
- Evidence for a dominant-negative effect in ACTA1 nemaline myopathy caused by abnormal folding, aggregation and altered polymerization of mutant actin isoforms / B. Ilkovski, K.J. Nowak, A. Domazetovska, A.L. Maxwell, S. Clement, K.E. Davies, N.G. Laing, K.N. North, S.T. Cooper // Hum. Mol. Genet. 2004. Vol. 13, No 16. P. 1727-1743. DOI: 10.1093/hmg/ddh185.
- Прогрессирующая немалиновая миопатия у пациентки, неоднократно оперированной на позвоночнике: клинический случай и обзор литературы / О.Г. Прудникова, А.О. Котельников, Н.С. Мигалкин, Г.Н. Филимонова // Хирургия позвоночника. 2022. T. 19, № 1. С. 15-21. DOI: 10.14531/ss2022.1.15-21.
- Lan T.H., Huang X.Q., Tan H.M. Vascular fibrosis in atherosclerosis // Cardiovasc. Pathol. 2013. Vol. 22, No 5. P. 401-407. DOI: 10.1016/j. carpath.2013.01.003.
- Manon-Jensen T., Kjeld N.G., Karsdal M.A. Collagen-mediated hemostasis // J. Thromb. Haemost. 2016. Vol. 14, No 3. P. 438-448. DOI: 10.1111/ jth.13249.
- Vascular Smooth Muscle Cells and Arterial Stiffening: Relevance in Development, Aging, and Disease / P. Lacolley, V. Regnault, P. Segers, S. Laurent // Physiol. Rev. 2017. Vol. 97, No 4. P. 1555-1617. DOI: 10.1152/physrev.00003.2017.
- Neuromuscular Disease Center. Washington University, St. Louis, MO USA. 2021. URL: http://neuromuscular.wustl.edu/index.html (дата обращения: 11.02.2022).
- Impairments in contractility and cytoskeletal organization cause nuclear defects in nemaline myopathy / J.A. Ross, Y. Levy, M. Ripolone, J.S. Kolb, M. Turmaine, M. Holt, J. Lindqvist, K.G. Claeys, J. Weis, M. Monforte, G. Tasca, M. Moggio, N. Figeac, P.S. Zammit, H. Jungbluth, C. Fiorillo, J. Vissing, N. Witting, H. Granzier, E. Zanoteli, E.C. Hardeman, C. Wallgren-Pettersson, J. Ochala // Acta Neuropathol. 2019. Vol. 138, No 3. P. 477495. DOI: 10.1007/s00401-019-02034-8.
- In vivo characterization of skeletal muscle function in nebulin-deficient mice / C. Gineste, A.C. Ogier, I. Varlet, Z. Hourani, M. Bernard, H. Granzier, D. Bendahan, J. Gondin // Muscle Nerve. 2020. Vol. 61, No 3. P. 416-424. DOI: 10.1002/mus.26798.
- Muscle histopathology in nebulin-related nemaline myopathy: ultrastructructural findings correlated to disease severity and genotype / E. Malfatti, V.L. Lehtokari, J. Böhm, J.M. de Winter, U. Schäffer, B. Estournet, S. Quijano-Roy, S. Monges, F. Lubieniecki, R. Bellance, M.T. Viou, A. Madelaine, B. Wu, A.L. Taratuto, B. Eymard, K. Pelin, M. Fardeau, C.A. Ottenheijm, C. Wallgren-Pettersson, J. Laporte, N.B. Romero // Acta Neuropathol. Commun. 2014. Vol. 2. P. 44. DOI: 10.1186/2051-5960-2-44.
- Бакланов А.Н., Колесов С.В., Шавырин И.А. Оперативное лечение нейромышечного сколиоза // Гений ортопедии. 2013. № 2. C. 72-77.
- Снижение периоперационного риска при вертебрологических операциях у пациентов с наследственными заболеваниями соединительной ткани / С.О. Рябых, В.Л. Шушарина, П.В. Очирова, А.Н. Третьякова, Т.В. Рябых // Гений ортопедии. 2015. № 4. C. 48-52. DOI: 10.18019/10284427-2015-4-48-52.
