Определение оптимальных параметров моделирования для максимально точных расчётов энергий в ОЦК-железе

Бесплатный доступ
В новой версии (11.1) программного пакета WIEN2k проведено первопринципное моделирование равновесной структуры и свойств ОЦК-железа. Определены оптимальные значения основных параметров моделирования, позволяющие рассчитывать энергетические характеристики системы с точностью не менее 0,01 эВ. Энергия растворения углерода в ОЦК-железе составила 0,85 эВ.
Оцк-железо, парамагнитное состояние, первопринципное моделирование, примеси углерода, метод lapw
Короткий адрес: https://sciup.org/147158923
IDR: 147158923 | УДК: 669.112.227.1:538.915 | DOI: 10.14529/mmph160408
Determining the optimal modeling parameters for maximum precise calculations of energy in BCC-iron
The ab initial modeling of the equilibrium structure and properties of BCC-iron is performed in the new version (11.1) of WIEN2k software package. A full-potential method of linear joined plane waves LAPW is used for the calculations, taking into account the generalized gradient approximation PBE-GGA, in supercell consisted of 54 iron atoms with periodic boundary conditions. This is the most accurate method used in the framework of density functional theory. Integration into the reciprocal space and calculation of electron density is held in accordance with the Monkhorst-Pack scheme with a grid of Nk points in the Brillouin zone. The criterion for the convergence in all the calculations is to achieve the accuracy of the calculation of the total energy of the system, charge and force of interaction between two atoms of not less than 10-4 Ry, 10-3е and 1 mRy/a.u. respectively. The optimal values of the basic simulation parameters of carbon impurities in the BCC-iron are determined. They allow calculating the energy performance of the system with an accuracy of not less than 0,01 eV. These parameters compile Nk = 64 points, Kmax = 5 a.u.-1. It is shown that the use of the simulation parameters obtained in previous versions of WIEN2k leads to the error in determining the carbon dissolving power in BCC-iron at 0,07 eV. The calculation of energy of dissolution of carbon atoms in the ferromagnetic phase of BCC-iron is conducted using the obtained simulation parameters. It amounts to 0,85±0,01 eV, which is a good fit to the experimental results and other first-principle calculations.
Текст научной статьи Определение оптимальных параметров моделирования для максимально точных расчётов энергий в ОЦК-железе
Сплавы на основе железа, несмотря на развитие производства других металлов, пластмасс, керамики, композитов, остаются основными конструкционными материалами. Первопринципное моделирование ОЦК-железа проводилось неоднократно, существует множество работ по этой теме [1–9]. В работах [1–4] моделирование проводилось псевдопотенциальным методом, который точно описывает электронный спектр только валентных электронов. Этого вполне достаточно для описания чистых веществ, однако может приводить к погрешностям при расчете растворов внедрения, где может возникнуть необходимость в учете более глубоких электронных уровней. Отсутствие учёта этих уровней приводит к менее точным результатам. В отличие от псевдопо-тенциального метода в полнопотенциальном происходит учёт всех электронов, что делает его наиболее точным. Данный метод используется в программном пакете WIEN2k [5–9]. В большинстве работ [7–9] отсутствует объяснение причины выбора тех или иных значений параметров моделирования. Задача выбора оптимальных параметров системы для моделирования электронных и магнитных свойств является чрезвычайно важной, поскольку от них зависит точность полученных результатов. Частично это связано с тем, что ряд параметров моделирования оптимизированы самими разработчиками пакета WIEN2k, либо группами, которые тесно с ними сотрудничают. Для расчетов сплавов на основе железа обычно используют обобщенное градиентное приближение PBE-GGA и типовые значения параметров, характеризующие базис плоских волн, которые составляющие E cut = –7 Рб, G max = 20 Рб0,5 [10–12]. Другие параметры моделирования были оптимизированы в работах нашей группы [5–6] и составили: Nk = 3x3x3 точки; Kmax = 5 а.е.–1; R mt (Fe) = 2,0 а.е.
В версии WIEN2k_11.1 [13] произошла смена схемы расчёта полной энергии системы. Смена расчётной схемы позволила получить более быструю и монотонную сходимость. Можно предполагать, что такое изменение приведет к тому, что параметры моделирования, оптимизированные для предыдущих версий программы, должны быть переопределены. Повышение достоверности и точности прогнозирования структуры и свойств, как чистого железа, так и сплавов на его основе является насущной задачей, стоящей перед материаловедами. В связи с этим настоящая работа посвящена выбору оптимальных параметров для построения модели ОЦК-железа в новой версии программного пакета WIEN2k, обеспечивающих точность расчета полной энергии моделируемой системы не хуже 0,01 эВ.
Методика расчётов
В данной работе расчёты проводились из первых принципов методом LAPW [10] в программном пакете WIEN2k [11], обеспечивающим высокую точность расчета полной энергии при минимальном количестве подгоночных параметров. Это наиболее точный метод, используемый в рамках теории функционала плотности. Для расчётов использовался вычислительный комплекс
Физика
ЮУрГУ-Торнадо [14]. Для интегрирования в обратном пространстве и вычисления электронной плотности использовалась схема Монхорста–Пака с сеткой из Nk точек в зоне Бриллюэна. Чем больше используется точек, тем выше точность интегрирования, однако при этом возрастает расчетное время, поэтому оптимальное значение определяется из условия достижения требуемой точности энергии системы [15]. Критерием сходимости во всех расчётах было достижение точности расчета полной энергии системы, заряда и силы взаимодействия между двумя атомами не менее 10–4 Рб, 10–3 е и 1 мРб/а.е. соответственно. В данной работе ОЦК-железо моделировалось в суперячейке состоящей из 54 атомов железа.
Для определения энергии растворения углерода относительно фазы графита использовалась формула:
∆ H = E (Fe 54 C) - E (Fe 54 ) - E (C), (1) где E (Fe 54 C) – энергия суперячейки, состоящей из 54 атомов железа и одного атома углерода, находящегося в октапоре, E (Fe 54 ) – энергия суперячейки, состоящей из 54 ат. железа, а E (C) – энергия одного атома углерода в решётке графита. Параметры структуры графита были взяты из работы Джианг [3] ( a = 2,462 Å, c = 6,656 Å, α = 90°, β = 90°, γ = 120°).
Результаты
В данной работе проведены вычисления, с помощью которых определены оптимальные параметры, позволяющие обеспечить высокую точность расчетов с приемлемыми затратами компьютерных ресурсов. Часть параметров можно взять из нашей работы [16]. В ней моделировалось растворение углерода в ГЦК-железо на 11.1 версии WIEN2k. Эти параметры R mt (Fe) = 2,0 а.е. и R mt (C) = 1,2 а.е. Значение R mt (Fe) = 2,0 а.е. совпало со значением полученным в работах нашей группы на предыдущих версиях WIEN2k [5–6].
В первую очередь была выполнена оптимизация количества k -точек, так как этот параметр не зависит от других. Были взяты значения, задаваемые для ОЦК-железа по умолчанию: a = 2,84 Å, K max = 5,0 а.е.–1 [5–6]. На рис. 1 представлен график зависимости энергии системы от количества Nk точек в зоне Бриллюэна.
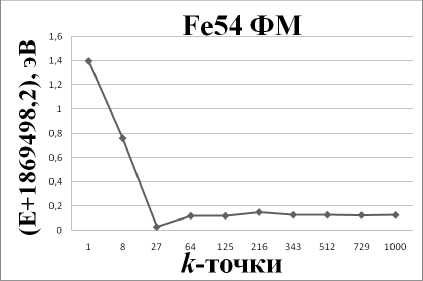
Рис. 1. График зависимости энергии системы от количества k -точек в зоне Бриллюэна
Из рис. 1 видно, что при увеличении количества k -точек выше 64 энергия системы перестаёт меняться. Таким образом, оптимальное число k -точек, необходимое для получения требуемой точности расчета полной энергии для суперячейки из 54 атомов, равно 64. Несимметричные k -сетки решено не рассматривать, ввиду того что система у нас симметричная и у неё нет никаких выделенных направлений для растяжения.
Весьма важным параметром моделирования является величина K max. Для оптимизации данного параметра была изучена сходимость результатов расчета полной энергии системы от K max , для ферромагнитной (ФМ) фазы ОЦК-железа, представленная на рис. 2. Видно, что в новой версии пакета, в отличии от прежней, не наблюдается сходимость значений энергии при значении параметра K max = 5 а.е.–1.
Расчеты показали, что с увеличением K max энергия системы монотонно падает, в силу чего не удается достичь такого значения параметра, после которого наблюдается сходимость в пределах 0,01 эВ для полной энергии системы. Отметим, однако, что реальной величиной, которую мы хотим получить при проведении расчетов, является не сама энергия, а ее разность для двух раз-
Ридный Я.М., Мирзоев А.А., Определение оптимальных параметров моделирования
Мирзаев Д.А. для максимально точных расчётов энергий в ОЦК-железе личных конфигураций суперячейки. В работе [6], например, проводилась оптимизация Kmax для достижения заданной точности расчета энергии растворения водорода в ОЦК-железе и было рекомендовано значение Kmax = 5 а.е.–1. Следуя примеру работы [6], мы решили определить оптимальное значение данного параметра по сходимости расчетной величины энергии растворения атома углерода в железе. С этой целью нами были определены равновесные значения параметров решётки для чистого железа и железа с одним атомом углерода (рис. 3).
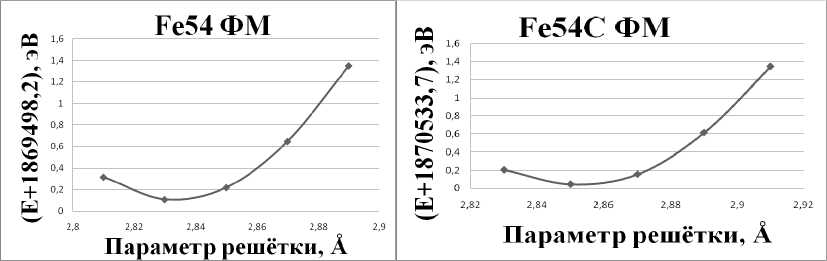
Рис. 3. График зависимости энергии системы от параметра решётки для чистого железа и железа с одним атомом углерода
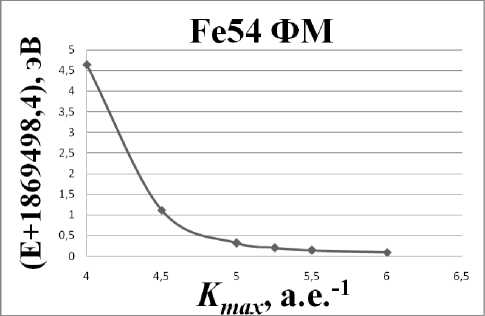
Рис. 2. График зависимости энергии системы от параметра K max
Из рис. 3 видно, что оптимальные параметры решётки a = 2,835 Å для чистого железа и a = 2,85 Å для системы с одним растворённым атомом углерода. Параметр решётки чистого железа практически совпадает с параметром определённым в работе [5], a = 2,84 Å. Небольшие отличия в 0,2 % являются следствием изменений схемы расчёта.
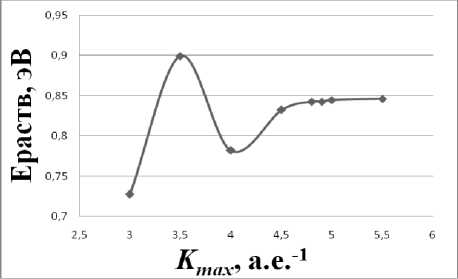
Рис. 4. График зависимости энергии растворения углерода от параметра K max
После определения равновесных параметров решётки, которые мало чувствительны к выбору параметра K max , была изучена сходимость значений по энергии растворения углерода в ОЦК -железе при вариации этого параметра (рис. 4). На графике видно, что наименьшим (оптимальным) значением параметра, обеспечивающим точность значений энергии не хуже 0,01 эВ, является K max = 5 а.е.–1. Таким образом, определенные нами оптимальные параметры моделирования
Физика
совпадают с параметрами, полученными в работах [5–6], за исключением числа k -точек, число которых необходимо увеличить с 27 до 64 точек.
После определения оптимальных параметров моделирования проведено сравнение энергии растворения углерода в ОЦК-железе, полученной в нашей работе, со значениями, полученными другими авторами (см. таблицу ) .
Энергия растворения углерода в ОЦК-железе
|
Работа |
Метод |
Энергия растворения, эВ |
|
[3,4] |
Псевдопот. |
0,7–0,74 |
|
[17] |
Терм. анализ |
0,99 |
|
[18] |
Эксп. ( Т = 1000 К) |
1,01 |
|
[19] |
Эксп. ( Т = 955–1000 К) |
1,14–1,19 |
|
[20] |
Эксп. ( Т = 773–993К) |
0,6–0,78 |
|
[21] |
Эксп. ( Т = 873–1073 К) |
0,63–0,98 |
|
Данная работа |
Полнопот. (27 k -точки) |
0,92±0,01 |
|
Данная работа |
Полнопот. (64 k -точки) |
0,85±0,01 |
Из таблицы видно, что данные по энергии растворения очень сильно различаются. Вычисленное нами значение энергии растворения углерода в ОЦК-железе лежит в середине интервала энергий растворения вычисленных экспериментально, что свидетельствует о более высокой точности проведенного моделирования, чем в работах [3, 4]. Показано, что использование параметров моделирования полученных в предыдущих версиях WIEN2k приводит к ошибке в определении энергии растворения углерода в ОЦК-железе в 0,07 эВ.
Заключение
Определены оптимальные значения основных параметров моделирования примесей углерода в ОЦК-железе, позволяющие рассчитывать энергетические характеристики системы с точностью не менее 0,01 эВ в новой версии (11.1) программного пакета WIEN2k. Показано, что:
-
1. Для обеспечения данной точности необходимо увеличить количество k -точек до 64 по сравнению со значением k = 27, рекомендованным в работах [5, 6]. Оптимальное значение параметра Kmax не изменилось и по-прежнему составляет 5 а.е.–1.
-
2. С использованием найденных значений параметров моделирования проведено вычисление энергии растворения атома углерода в ферромагнитной фазе ОЦК-железа, которая составила 0,85 эВ, что находится в хорошем согласии с результатами эксперимента и других первоприн-ципных расчетов. Использование параметров моделирования, полученных для предыдущих версий WIEN2k, приводит к ошибке в определении энергии растворения углерода в ОЦК-железе в 0,07 эВ.
Исследование выполнено за счет гранта Российского научного фонда № 16-19-10252 и гранта РФФИ № 16-03-00486 А.
Список литературы Определение оптимальных параметров моделирования для максимально точных расчётов энергий в ОЦК-железе
- Domain, C. Ab initio Study of Foreign Interstitial Atom (C, N) Interactions with Intrinsic Point Defects in α-Fe/C. Domain, C.S. Becquart, J. Foct//Physical Review B. -2004. -Т. 69, № 14. -P. 144112.
- Ruban, A.V. Self-trapping of carbon atoms in a-Fe during the martensitic transformation: A qualitative picture from ab initio calculations/A.V. Ruban//Physical Review B. -2014. -Vol. 90. -P. 144106.
- Jiang, D.E. Carbon dissolution and diffusion in ferrite and austenite from first principles/D.E. Jiang, E.A. Carter//Physical Review B. -2003. -Vol. 67. -P. 214103.
- Hepburn, D.J. First-principles study of helium, carbon, and nitrogen in austenite, dilute austenitic iron alloys, and nickel/D.J. Hepburn, D. Ferguson, S. Gardner, G.J. Ackland//Physical Review B. -2013. -Vol. 88. -P. 024115.
- Урсаева, А.В. Выбор оптимальных параметров для построения максимально точной модели ОЦК-железа/А.В. Урсаева, Г.Е. Рузанова, А.А. Мирзоев//Вестник ЮУрГУ. Серия «Математика. Механика. Физика». -2010. -Вып. 2, № 9(185). -С. 97-101.
- Ракитин, M.C. Изменение электронной структуры α-железа, содержащего внедренные атомы водорода/M.C. Ракитин, А.А. Мирзоев, Д.А. Мирзаев//Вестник ЮУрГУ. Серия «Металургия». -2010. -Вып. 14, № 13(189). -C. 67-71.
- Stojica, N.L. Phase stability of Fe and Mn within density-functional theory plus on-site Coulomb interaction approaches/N.L. Stojica, N.L. Binggeli//Journal of Magnetism and Magnetic Materials. -2008. -Vol. 320. -P. 100-106.
- Iglesias, R. Ab initio studies on the magnetic phase stability of iron/R. Iglesias, S.L. Palacios//Acta Materialia. -2007. -Vol. 55. -P. 5123-5129.
- Herper, H.C. Ab initio full-potential study of the structural and magnetic phase stability of iron/H.C. Herper, E. Hoffmann, P. Entel//Physical Review B. -1999. -Vol. 60. -P. 3839-3848.
- Cottenier, S. Density Functional Theory and the family of (L)APW-methods: a step-by-step introduction/S. Cottenier, 2004.
- Schwarz, К. Electronic structure calculations of solids using the WIEN2k package for material science/K. Schwarz, P. Blaha, G.K.H. Madsen//Computer Physics Communications. -2002. -Vol. 147. -P. 71-76.
- Blaha, P. An Augmented PlaneWave + Local Orbitals Program for Calculating Crystal Properties revised edition WIEN2k_11.1 (Release 5.4.2011)/P. Blaha, K. Schwarz, G. Madsen et. al. .
- Marks, L.D. Fixed-Point Optimization of Atoms and Density in DFT/L.D. Marks//J. Chem. Theory Comput. -2013. -Vol. 9, № 6. -P. 2786-2800.
- http://supercomputer.susu.ac.ru/computers/tornado/
- Monkhorst, H.J. Special points for Brillouin-zone integrations/H.J. Monkhorst, J.D. Pack//Physical Review B. -1976. -Vol. 13, № 12. -P. 5188-5192.
- Ридный, Я.М. Ab-initio моделирование влияния ближнего окружения примесей углерода на энергию их растворения в ГЦК-железе/Я.М. Ридный, А.А. Мирзоев, Д.А. Мирзаев//Вестник ЮУрГУ. Серия «Математика. Механика. Физика». -2013. -Т. 5, № 2. -С. 108-116.
- Могутнов, Б.М. Термодинамика сплавов железа/Б.М. Могутнов, И.А. Томилин, Л.А. Шварцман. -Москва: Металлургия, 1984. -206 с.
- Dunn, W.W. The thermodynamic properties of carbon in body-centered cubic iron/W.W. Dunn, R.B. McLellan//Metallurgical Transactions. -1971. -Vol. 2. -P. 1079-1086.
- Lobo, J.A. Thermodynamics and solubility of carbon in ferrite and ferritic Fe-Mo alloys/J.A. Lobo, G.H. Geiger//Metallurgical Transactions A. -1976. -Vol. 7A. -P. 1347-1357.
- Shumilov, M. Solubility of carbon in ferrite/M. Shumilov, A. Kozak, L. Yakushechkina, K. Sokolov//The Physics of Metals and Metallography. -1973. -Vol. 47. -P. 2169-2178.
- Schlirmann, E. Carburisation equilibria of alpha-iron with methane-hydrogen mixtures in the 600-800 C range and their/E. Schlirmann, T. Schmidt, F. Tillmann//Giesserei-Forschung. -1967. -Vol. 19, № 1. -pp. 35-41.
