Определение содержания активного компонента экстракта аврана лекарственного (кукурбитацинa B) методом высокоэффективной жидкостной хроматографии с масс-спектрометрическим детектированием при введении экстракта лабораторным животным
 методом высокоэффективной жидкостной хроматографии с масс-спектрометрическим детектированием при введении экстракта лабораторным животным")
Автор: Фомина Ю.А., Шестопалова Н.Б., Калюта Т.Ю., Кенжегулова А.А., Попов Н.С., Сивас И.С., Федонников А.С.
Журнал: Саратовский научно-медицинский журнал @ssmj
Рубрика: Фармакология, клиническая фармакология
Статья в выпуске: 4 т.21, 2025 года.
Бесплатный доступ
Цель: разработка и валидация хроматомасс-спектрометрической методики количественного определения кукурбитацина B в плазме крови и гомогенатах печени, почек и сердца мышей и оценка содержания и динамики концентрации кукурбитацина В в крови животных при однократном пероральном введении. Материал и методы. Изучение фармакокинетики выполнено на 60 беспородных мышах мужского пола массой 28,0±3,0 г. Животные получали экстракт аврана лекарственного сухой в виде водного раствора. Для разработки методики количественного определения кукурбитацина В использовали высокоэффективный жидкостной хроматограф Agilent Technologies 1260 Infinity II и масс-спектрометр AB Sciex QTrap 3200 MD. Хроматографическое разделение осуществляли на аналитической колонке Agilent InfinityLab Poroshell 120 EC-C18 с градиентной подвижной фазой. Пробоподготовка плазмы крови мышей заключалась в осаждении белков метанолом. Результаты. На основании результатов определения кукурбитацина В в плазме крови мышей после однократного внутрижелудочного введения аврана лекарственного экстракта в дозе 220,0 мг/кг выявлено, что во всех анализируемых образцах уровень сигнала находится ниже уровня предела количественного измерения. Заключение. Разработана биоаналитическая методика определения кукурбитацина В в плазме крови мышей с помощью высокоэффективной жидкостной хроматографии с масс-спектрометрическим детектированием, подобраны параметры идентификации кукурбитацина В в плазме крови мышей после однократного внутрижелудочного введения аврана лекарственного экстракта.
Кукурбитацин B, аврана лекарственного экстракт, фармакокинетика, масс-спектрометрия, высокоэффективная жидкостная хроматография
Короткий адрес: https://sciup.org/149150227
IDR: 149150227 | УДК: 615.03:615.038 | DOI: 10.15275/ssmj2104488
Determination of the content of the active component of Gratiola officinalis L. extract cucurbitacin B by liquid chromatography with tandem mass spectrometry after administation to laboratory animals
Objective: to develop and validate a chromatograph mass spectrometric method for the quantitative determination of cucurbitacin B in mouse plasma and liver, kidney, and heart homogenates, and to evaluate the blood cucurbitacin B levels and concentration dynamics following a single oral administration. Material and methods. An Agilent Technologies 1260 Infinity II high-performance liquid chromatograph and an AB Sciex QTrap 3200 MD mass spectrometer were used to develop the method for the quantitative determination of cucurbitacin B. Chromatographic separation was performed on an Agilent InfinityLab Poroshell 120 EC-C18 analytical column with a gradient mobile phase. Mouse plasma sample preparation consisted of protein precipitation with methanol. Results. To study the pharmacokinetics of the components of the Grapevine officinalis extract, a bioanalytical method for determining cucurbitacin B in mouse plasma using high performance liquid chromatography with tandem mass spectrometry was developed, and cucurbitacin B identification parameters were selected (lower limit of quantification). Conclusion. To determine the pharmacokinetic parameters of cucurbitacin B, it is necessary to search for other analytes – cucurbitacin B metabolites. Consequently, it is impossible to construct a pharmacokinetic curve for cucurbitacin B. The metrological characteristics of the method allow it to be used for the analytical portion of pharmacokinetic studies of both individual substances and plant extracts.
Текст научной статьи Определение содержания активного компонента экстракта аврана лекарственного (кукурбитацинa B) методом высокоэффективной жидкостной хроматографии с масс-спектрометрическим детектированием при введении экстракта лабораторным животным
EDN: UYWJUO
биологических жидкостях для оценки фармакокинетики данного компонента при его применении in vivo .
Цель – разработка и валидация хроматомасс-спектрометрической методики количественного определения кукурбитацина B в плазме крови и гомогенатах печени, почек и сердца мышей и оценка содержания и динамики концентрации кукурбитаци-на В в крови животных при однократном пероральном введении.
Материал и методы . Изучение фармакокинетики выполнено на 60 беспородных мышах мужского пола массой 28,0±3,0 г. Животные получали экстракт аврана лекарственного сухой в виде водного раствора в дозе 220,0 мг/кг. Из таблеток, содержащих аврана лекарственного экстракт, готовили формуля-цию следующим образом: 6 таблеток с дозировкой экстракта аврана сухого 125 мг помещали в коническую стеклянную колбу (100 мл) и добавляли деионизированную воду в объеме 91 мл, перемешивали на шейкере до полного растворения. Таким образом, полученная формуляция содержала в 1 мл 8,24 мг экстракта, что при введении мышам массой 30,0 г 0,8 мл формуляции доза составила 220,0 мг/кг. При выполнении эксперимента соблюдались все условия, предусмотренные Хельсинской декларацией и ее редакцией (2024) и Положением об использовании животных в биомедицинских исследованиях, принятым 41-й Генеральной ассамблеей Всемирной медицинской ассоциации (1989), в которых регламентированы этические нормы проведения экспериментов на животных. При работе с экспериментальными животными руководствовались требованиями ГОСТа 33215–2014 «Руководство по содержанию и уходу за лабораторными животными. Правила оборудования помещений и организации процедур»и ГОСТа 33216– 2014 «Руководство по содержанию и уходу за лабораторными животными. Правила содержания и ухода за лабораторными грызунами и кроликами».
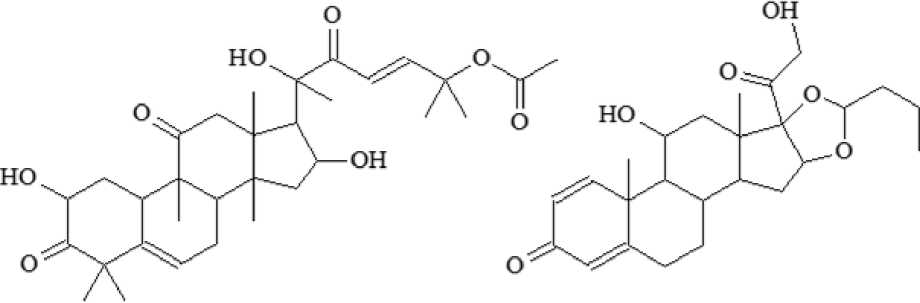
Разработана и валидирована методика определения кукурбитацина В в биологических образцах лабораторных животных. В процессе разработки методики использовали аналитический стандарт кукурбитаци-на B (PhytoLab, Германия). В качестве внутреннего стандарта (IS) применяли будесонид («АстраЗенека АБ», Швеция; рис. 1).
В процессе хроматографического анализа и про-боподготовки образцов использовали следующее оборудование: аналитические весы ВЛ-124 («Гос-метр», Россия), одноканальные автодозаторы Research Plus (Eppendorf AG, Германия), центрифу-гу-вортекс Microspin FV-2400 (Biosan, Латвия), термостатируемый шейкер TS-100C (Biosan, Латвия), центрифугу с охлаждением Sigma 1-14K (Sigma, Германия), морозильный шкаф -40ºC MDF-136 (Sanyo, Япония), холодильник +4ºC с морозильной камерой -20ºC GA-B409UCA (LG, КНР).
а б
Рис. 1. Структурные формулы кукурбитацина B ( a ) и будесонида ( б )
Плазму мышей получалипутем центрифугирования цельной гепаринизированной крови при 3000 об/мин в течение 10 мин. Гомогенаты внутренних органов (печени, почек, сердца) получали следующим образом: фрагмент каждой ткани промывали в физиологическом растворе, обсушивали фильтровальной бумагой и помещали в предварительно взвешенные на аналитических весах пробирки типа Эппендорф объемом 2 мл. Определяли массу фрагмента ткани путем повторного взвешивания заполненной пробирки. Немедленно добавляли деионизированную воду из расчета на 100 мг ткани – 400 мкл. В пробирку добавляли бусину из кварцевого стекла диаметром 5 мм и подвергали колебаниям амплитудой 30 мм и частотой 50 Гц до полной гомогенизации.
Для изготовления 10-кратных рабочих растворов (серия разведений на ацетонитриле [Scharlau, Испания]) кукурбитацина B готовили исходный раствор на метаноле в концентрации 0,5 мг/мл. Рабочие растворы были использованы для приготовления калибровочных (k) и контрольных образцов (QC) на плазме крови и гомогенатах печени, почек и сердца мышей. Раствор IS (будесонида) готовили путем растворения суспензии (250 мкг/мл) в метаноле до достижения концентрации 5 мкг/мл.
С помощью высокоэффективного жидкостного хроматографа 1260 Infinity II (Agilent Technologies, Германия) в обращенно-фазовом режиме с использованием аналитической колонки Poroshell InfinityLab 120 EC-C18 4,6×100 мм, 2,7 мкм (Agilent Technologies, США) в сочетании с предколонкой Zorbax Eclipse Plus C18 4,6×12,5 мм, 5 мкм (Agilent Technologies, США) проводили количественное определение кукурбита-цина B . Элюирование осуществляли смесью деионизированной воды и ацетонитрила с добавлением 0,1% муравьиной кислоты (Fischer, Германия) в градиентном режиме.
Детектирование кукурбитацина B и будесонида при проведении хроматографического анализа осуществляли с помощью масс-спектрометра типа тройной квадруполь AB Sciex QTrap 3200MD (AB Sciex Pte. Ltd., Сингапур) с электрораспылительным источником ионов. Подбор оптимальных параметров детекции проводили при непрерывном введении растворов кукурбитацина B и будесонида (растворитель – 50% метанол с добавлением 0,1% муравьиной кислоты) в источник ионов с помощью шприцевого насоса со скоростью 10 мкл/мин. На I этапе для аналита и внутреннего стандарта определяли массовое число (m/z) депротонированных молекул или соответствующих аддуктов, подбирали оптимальные значения потенциала декластеризации (DP) и напряжения на входе в ячейку соударений (CEP). Для детекции кукурбитацина B и будесонида в режиме мониторинга множественных реакций (MRM) и обеспечения наилучшей чувствительности на II этапе работы подбирали оптимальные значения энергии столкновений (CE) и ускоряющего напряжения (CXP) для установленных ионов-предшественников определяли масс-спектры второго порядка, выбирали характеристические ионы.
Пробоподготовку калибровочных и контрольных образцов осуществляли следующим образом: 90 мкл пулированной интактной плазмы крови или гомогената ткани мышей переносили в пробирки объемом 1,5 мл, добавляли 10 мкл соответствующего 10-кратного рабочего раствора кукурбитацина B и перемешивали на вортексе в течение 30 секунд. Затем добавляли 400 мкл раствора будесонида в метаноле (300 нг/ мл), предварительно охлажденного до -10ºС, перемешивали на термостатируемом шейкере (+4ºС) в течение 2 мин. Пробы центрифугировали в течении 15 мин при 15 000 g, 50 мкл супернатанта переносили в полиэтиленовые вставки в хроматографические виалы и использовали для последующего анализа посредством высокоэффективной жидкостной хроматографии (ВЭЖХ) с тандемной масс-спектрометрией (МС/МС) – ВЭЖХ-МС/МС. Пробоподготовку исследуемых образцов осуществляли согласно с соответствующей методикой для калибровочных и контрольных образцов.
Первичные данные хроматомасс-спектрометрического анализа обрабатывали с помощью программного обеспечения AB Sciex Analyst 1.6.3 (AB Sciex, США), для расчета значений валидационных параметров использовали пакет программ Microsoft Office Excel 365 (Microsoft Corp., США).
Разработанную методику валидировали по следующим параметрам: селективность, матричный эффект, перенос пробы, линейность аналитического диапазона, нижний предел количественного определения (lower limit of quantification – LLOQ), внутри-и межсерийная точность и прецизионность, стабильность на всех этапах анализа.
Концентрации аналитов рассчитаны с помощью программного обеспечения Analyst 1.6.3 по калибровочным графикам зависимости площади хроматографического пика аналита, нормированной на площадь IS, от номинальной концентрации аналита.
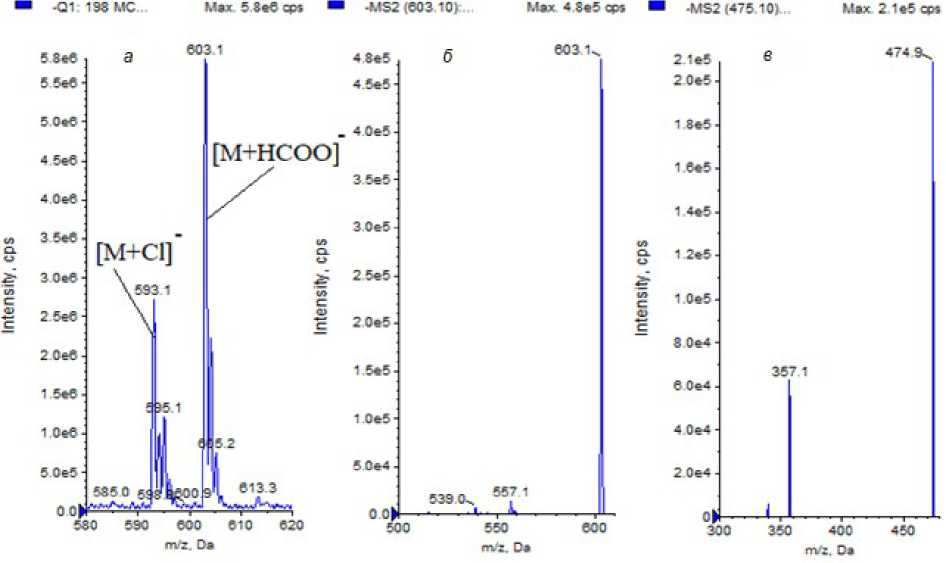
Рис. 2. Масс-спектры первого порядка аддуктов кукурбитацина B ( a ) и второго порядка – для кукурбитацина B ( б ) и будесонида ( в ) в режиме регистрации отрицательных ионов
Статистическая обработка данных проведена в программе Microsoft Office Excel 365. Рассчитывали среднее арифметическое значение, его стандартное отклонение, коэффициент вариации. В работе использован регрессионный анализ.
Результаты. На I этапе исследования для кукур-битацина B и будесонида получены масс-спектры первого и второго порядка, значения их m/z фрагментарных ионов сравнивали с данными литературы. Для аналита и внутреннего стандарта выбраны характеристические ионы-продукты, для которых были подобраны условия детектирования, обеспечивающие максимальный ионный ток (табл. 1). Масс-спектр ионов-предшественников аддуктов кукур-битацина B представлен на рис. 2, а , масс-спектры второго порядка для аналита и внутреннего стандарта представлены на рис. 2, б и в соответственно.
Хроматографическое определение кукурбитаци-на B осуществляли в обращенно-фазовом режиме смесью деионизированной воды (рис. 2, а ) и ацетонитрила (рис. 2, б ) с добавлением 0,1% муравьиной кислоты. Программа градиента представлена в табл. 2. Время удерживания кукурбитацина B и будесонида (IS) составило 7,55 и 7,60 мин соответственно при общей продолжительности хроматографического анализа 10 мин.
Для определения метрологических характеристик разрабатываемой методики была приготовлена и проанализирована серия стандартных образцов с содержанием кукурбитацина B : 0,5; 1,0; 5,0; 10,0; 50,0; 100,0; 500,0; 1000,0 и 2000,0 нг/мл в пересчете на плазму крови или гомогенат ткани. На основании результатов анализа были построены калибровочные кривые, отражающие зависимость отношения площади пика ку-курбитацина B (аналита) к площади пика будесонида
Таблица 1
Параметры масс-спектрометрической детекции кукурбитацина B и будесонида в режиме MRM
|
Тип источника ионов |
TurboIonSpray |
|
Режим ионизации |
Отрицательный |
|
Температура источника ионов, ºC |
400,0 |
|
Напряжение источника ионов, В |
-4500,0 |
Давление, psi
|
газа завесы газа-распылителя газа-нагревателя |
20,0 40,0 50,0 |
||||||
|
Кукурбитацин B |
MRM, m/z |
Dwell, мсек |
DP, В |
EP, В |
CEP, В |
CE, эВ |
CXP, В |
|
603,1/603,1; 603,1/557,1 |
100 |
-65,0 |
-5,0 |
-22,0 |
-45,0; -36,0 |
-1,0; -3,8 |
|
|
Будесонид (IS) |
475,1/357,1 |
-64,0 |
-21,0 |
-20,0 |
-2,5 |
||
Таблица 2
Селективность ВЭЖХ-МС/МС методики кукурбитацина и IS (будесонида) в плазме крови и гомогенатах органов мышей
|
Вещество |
Средняя площадь пика |
Селективность, % |
|
|
в холостом образце ( N =6) |
образце LLOQ ( N =6) |
||
Плазма крови
|
Кукурбитацин B |
134,43 |
1210,83 |
10,81 |
|
Будесонид (IS) |
23,10 |
22850,00 |
0,10 |
|
Гомогенат печени |
|||
|
Кукурбитацин B |
25,48 |
1050,33 |
2,50 |
|
Будесонид (IS) |
18,90 |
21066,67 |
0,09 |
|
Гомогенат почек |
|||
|
Кукурбитацин B |
136,82 |
1050,33 |
13,15 |
|
Будесонид (IS) |
16,80 |
21066,67 |
0,08 |
|
Гомогенат сердца |
|||
|
Кукурбитацин B |
24,69 |
1097,83 |
2,71 |
|
Будесонид (IS) |
12,60 |
21916,67 |
0,06 |
Таблица 3
Матричный эффект при определении кукурбитацина B в плазме крови и гомогенатах органов мышей
|
Биологический материал и статистические параметры |
Площадь пика |
NMF |
||
|
аналита |
IS |
аналита/IS |
||
|
Плазма крови |
||||
|
Среднее значение |
||||
|
в solvent spike sample |
6902,00 |
17400,00 |
0,40 |
– |
|
post-spike sample |
6394,00 |
16720,00 |
0,38 |
0,97 |
|
SD |
424,89 |
370,14 |
0,03 |
0,08 |
|
CV , % |
6,65 |
2,21 |
8,04 |
8,04 |
|
Гомогенат печени |
||||
|
Среднее значение |
||||
|
в solvent spike sample |
6670,00 |
17400,00 |
0,38 |
– |
|
post-spike sample |
6222,00 |
16560,00 |
0,38 |
0,98 |
|
SD |
412,64 |
391,15 |
0,02 |
0,06 |
|
CV , % |
6,63 |
2,36 |
6,41 |
6,41 |
|
Гомогенат почек |
||||
|
Среднее значение |
||||
|
в solvent spike sample |
6526,00 |
17080,00 |
0,38 |
– |
|
post-spike sample |
6636,00 |
16640,00 |
0,40 |
1,04 |
|
SD |
60,25 |
391,15 |
0,01 |
0,03 |
|
CV , % |
0,91 |
2,35 |
2,46 |
2,46 |
|
Гомогенат сердца |
||||
|
Среднее значение |
||||
|
в solvent spike sample |
6942,00 |
17980,00 |
0,39 |
– |
|
post-spike sample |
6580,00 |
17500,00 |
0,38 |
0,97 |
|
SD |
351,92 |
452,77 |
0,02 |
0,06 |
|
CV , % |
5,35 |
2,59 |
6,08 |
6,08 |
Таблица 4
Степень извлечения кукурбитацина B из плазмы крови и гомогенатов органов мышей
|
Биологический материал и статистические параметры |
Площадь пика кукурбитацина B |
Степень извлечения, % |
|
|
pre-spike sample |
post-spike sample |
||
|
Плазма крови |
|||
|
Среднее значение ( N =6) |
6854,00 |
6394,00 |
107,44 |
|
SD |
681,56 |
424,89 |
11,00 |
|
CV , % |
9,94 |
6,65 |
10,24 |
|
Гомогенат печени |
|||
|
Среднее значение ( N =6) |
6806,00 |
6222,00 |
109,27 |
|
SD |
745,14 |
412,64 |
6,94 |
|
CV , % |
10,95 |
6,63 |
6,36 |
|
Гомогенат почек |
|||
|
Среднее значение ( N =6) |
6680,00 |
6636,00 |
100,64 |
|
SD |
780,54 |
60,25 |
11,42 |
|
CV , % |
11,68 |
0,91 |
11,35 |
|
Гомогенат сердца |
|||
|
Среднее значение ( N =6) |
6428,00 |
6580,00 |
97,92 |
|
SD |
281,55 |
351,92 |
6,80 |
|
CV , % |
4,38 |
5,35 |
6,94 |
(IS) от концентрации аналита в стандартном образце. Калибровочная зависимость представлена в виде уравнений линейной регрессии с нормированием 1/ x 2. За LLOQ принимали минимальную концентрацию ку-курбитацина B , которая может быть определена с точностью 80–120% и прецизионностью не более ±20%, при этом соотношение «сигнал/шум» на хроматограмме должно быть не менее 5:1.
Оценку селективности методики проводили путем анализа холостых проб (double blank) и проб с концентрацией кукурбитацина B 10,0 нг/мл, приготовленных на 6 индивидуальных образцах интактной плазмы крови и гомогенатов печени, почек и сердца мышей. Результаты определения кукурбитацина B и будесо-нида в холостых образцах (k0), холостых образцах c добавкой внутреннего стандарта и образцах LLOQ представлены в табл. 2. Средние значения селективности для аналита и внутреннего стандарта составили менее 15 и 0,1% соответственно, что свидетельствует о незначительном влиянии компонентов матрицы на количественное определение кукурбитацина B .
Оценку влияния матричного эффекта на определение кукурбитацина B проводили для концентрации 100 нг/мл в 6 индивидуальных образцах плазмы крови и гомогенатов органов мышей. Матричный эффект рассчитывали как отношение сигнала аналита в неэк-страгированном образце (post-spike sample) к усредненному значению сигнала аналита в чистом растворителе (solvent-spike sample, N=6). Нормированный матричный фактор (non-negative matrix factorization – NMF) рассчитывали как отношение нормированного на IS значения площади пика кукурбитацина B в биоматериале на усредненное значение отношения нормированного на будесонид значения площади пика аналита в стандартном растворе. Приемлемые значения относительного стандартного отклонения CV<15% для NMF (см. табл. 3) показывают, что используемый в качестве внутреннего стандарта будесонид эффективно компенсируют влияние матрицы на сигнал кукурбитацина B и что присутствие матричных компонентов не будет отражаться на точности определения при анализе реальных образцов.
Определение степени извлечения кукурбитацина B проводили для концентрации 100 нг/мл в 6 индивидуальных образцах плазмы крови и гомогенатов органов мышей. Степень извлечения была рассчитана как отношение сигнала в экстрагированном образце (pre-spike sample) к сигналу в неэкстрагированном образце (post-spike sample). Результаты определения степени извлечения представлены в табл. 4. Средняя степень извлечения аналитов находится в пределах 97,92–109,27%, максимальный разброс ( СV ) 11,35%, что свидетельствует о практически количественном извлечении кукурбитацина B из биоматериала.
Оценку внутри-и межсерийной точности и прецизионности определения кукурбитацина B проводили в 3 независимых сериях путем анализа 5 контрольных образцов для 4 уровней концентрации: 10,0 (LLQC); 22,5 (контрольный образец с низкой концентрацией аналита – LQC); 900,0 (контрольный образец со средней концентрацией аналита – MQC); 1800,0 (контрольный образец с высокой концентрацией аналита – HQC) нг/мл. Точность была определена как отношение рассчитанной концентрации в контрольном образце к номинальному значению, выраженное в процентах. Прецизионность определяли как CV между результатами определения кукурбитацина B в контрольных образцах, выраженное в процентах. Критериями приемлемости для концентрации на уровне LLOQ считали точность в диапазоне 80–120%, прецизионность – ±20%; для концентраций выше LLOQ – точность в диапазоне 85–115%, прецизионность – ±15%. Полученные результаты показывают, что значения межсерийной точности и прецизионности соответствуют указанным критериям (табл. 5).
Для оценки переноса веществ во время хроматографического анализа сравнивали хроматограммы холостых проб, которые проанализированы после
6-кратного ввода образца с высокой концентрацией кукурбитацина B (2000 нг/мл), с хроматограммами образцов с содержанием аналита на уровне LLQC. По результатам анализа установлено, что отношения площадей пиков в холостых образцах к площадям пиков в образцах LLQC был ниже максимально допустимого уровня (20% для аналита и 5% – для IS).
Результаты оценки стабильности подтвердили сохранность исходных и рабочих растворов кукурбита-цина B и будесонида в течение 1 мес. Контрольные образцы, полученные путем добавления к плазме
Таблица 5
Точность и прецизионность определения кукурбитацина B в плазме крови и гомогенатах органов мышей
Таким образом, на I этапе была разработана биоаналитическая ВЭЖХ-МС/МС-методика количественного определения кукурбитацина B в плазме крови и гомогенатах печени, почек и сердца мышей, которая полностью соответствует валидационным требованиям.
На II этапе данную методику применяли для оценки концентрации кукурбитацина B в крови мышей. На этом этапе проведена оценка содержания в крови лабораторных животных кукурбитацина B . На основании результатов определения кукурбитацина В в плазме крови мышей после однократного внутриже-лудочного введения аврана лекарственного экстракта в дозе 220,0 мг/кг выявлено, что во всех анализируемых образцах уровень сигнала находится ниже LLOQ. Полученные данные не позволяют оценить фармакокинетику компонентов экстракта аврана модельным методом. Однако наличие сигналов от анализируемых веществ LLOQ, но превышающие предел детекции и отсутствие таковых в холостых образцах позволяет предположить абсорбцию этих веществ из желудочно-кишечного тракта мышей.
Обсуждение. На основании результатов определения кукурбитацина В в плазме крови и гомогенатах почек, печени и сердца мышей после однократного внутрижелудочного введения аврана лекарственного экстракта в дозе 220,0 мг/кг выявлено, что во всех анализируемых образцах для всех аналитов уровень сигнала находится ниже LLOQ [19]. Полученные данные не позволяют оценить фармакокинетику компонентов экстракта аврана с использованием модельных методов. Однако наличие сигналов от анализируемых веществ ниже LLOQ, но выше условного предела детекции (соотношение «сигнал/шум» более 3:1) и отсутствие таковых в холостых образцах позволяет сделать предположение об абсорбции этих веществ из желудочно-кишечного тракта мышей. Вероятно, применение экстракта аврана у мышей в дозах, значительно превышающих 220,0 мг/кг, позволит получить результаты количественного определения кукурбита-цина В , пригодные для оценки фармакокинетики.
Так, анализ результатов скрининговой оценки содержания анализируемых компонентов экстракта ав-рана в плазме крови мышей через 1,5 ч после внутри-желудочного введения препарата в дозе 1000,0 мг/кг показал, что концентрации для кукурбитацина B в данных условиях не удалось достичь уровня хроматографического сигнала, превышающего таковой в образце LLOQ.
Заключение. Для изучения фармакокинетики компонентов экстракта аврана лекарственного разработана биоаналитическая методика определения кукурбитацина В в плазме крови мышей с помощью ВЭЖХ-МС/МС, подобраны параметры идентификации кукурбитацина В. Валидация включала определение внутрисерийной точности и прецизионности (в течение 1 аналитической серии c использованием 5 параллельных проб) и межсерийной точности и прецизионности (в течение 3 последовательных аналитических серий в разные дни). Для всех калибровочных стандартов и контрольных образцов отклонение от номинальной концентрации (точность) не превышало 15% (20% – для LLOQ). Значения внутри-и межсерийной прецизионности не превышали 15%, что демонстрирует приемлемую прецизионность метода ВЭЖХ-МС/МС. В рамках валидации также изучено влияние матрицы на определение и извлечение аналитов в плазме крови мышей путем анализа образцов, приготовленных в 5 различных источниках матрицы. Относительное стандартное отклонение нормированного матричного фактора (CV) и степени извлечения (Recovery) не превышало 15% для всех изученных матриц и аналитов, что свидетельствует об отсутствии существенного влияния матрицы на точность и прецизионность определения и извлечение аналитов.
LLOQ для кукурбитацина В составил 10,0 нг/мл, при этом диапазон определяемых концентраций 10,0– 2000,0 нг/мл.
Однако применение данной методики невозможно для количественного определения кукурбитацин B в крови в неизменном виде.
На основании результатов определения кукурби-тацина В в плазме крови мышей после однократного внутрижелудочного введения аврана лекарственного экстракта в дозе 220,0 мг/кг выявлено, что во всех анализируемых образцах для всех аналитов уровень сигнала находится ниже LLOQ. Полученные данные не позволяют оценить фармакокинетику компонентов экстракта аврана с использованием модельных методов. Вместе с тем наличие сигналов от анализируемых веществ ниже LLOQ, но выше условного предела детекции (соотношение «сигнал/шум» более 3:1) и отсутствие таковых в холостых образцах. Именно поэтому для определения фармакокинетических параметров кукурбитацина B необходим поиск других аналитов – метаболитов кукурбитацина B , и, как следствие, констатируется отсутствие возможности построения фармакокинетической кривой кукурбита-цина B для высоких концентраций кукурбитацина B при пероральном введении.
Метрологические характеристики методики позволяют использовать ее для проведения аналитической части фармакокинетических исследований как препаратов индивидуальных веществ, так и различных растительных экстрактов.
Вклад авторов. Все авторы сделали эквивалентный вклад в подготовку публикации.
